

Translate this page into:
An Interesting Case of Lineage Switch to Histiocytic Sarcoma in a Case of Diffuse Large B Cell Lymphoma
-
Received: ,
Accepted: ,
This article was originally published by Thieme Medical and Scientific Publishers Private Ltd. and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Lineage switch involves change in the phenotypic characteristics from one type to another. It is a rare phenomenon described in mature lymphoid neoplasms which transform to histiocytic/dendritic cell tumor, more commonly described in low-grade lymphoma like follicular, chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL), wherein the neoplasm loses the phenotypic characteristics of non-Hodgkin lymphoma and acquires the markers of histiocytic differentiation. Here, we present a case of diffuse large B cell lymphoma transforming to histiocytic sarcoma post 6 months of start of therapy. Histiocytic sarcoma being a very aggressive tumor, the patient had a very rapid deteriorating course and succumbed to disease.
Keywords
diffuse large B cell lymphoma
histiocytic sarcoma
lineage switch

Introduction
Lineage switch involves change in the phenotypic characteristics from one type to another. It is a rare phenomenon described in mature lymphoid neoplasms which transform to histiocytic/dendritic cell tumor, more commonly described in low-grade lymphoma like follicular, chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL), wherein the neoplasm loses the phenotypic characteristics of non-Hodgkin lymphoma (NHL) and acquires the markers of histiocytic differentiation.
Case Report
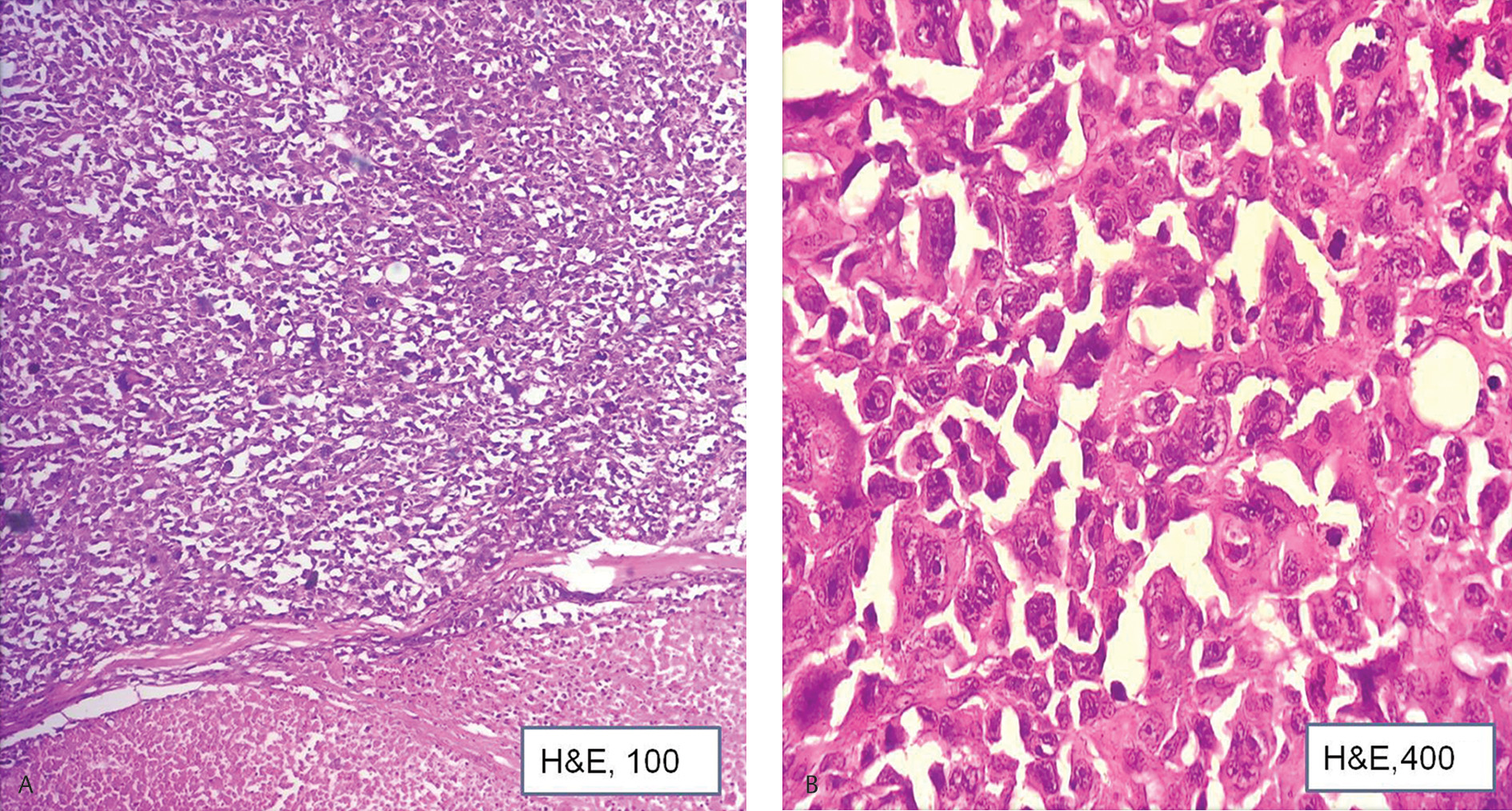
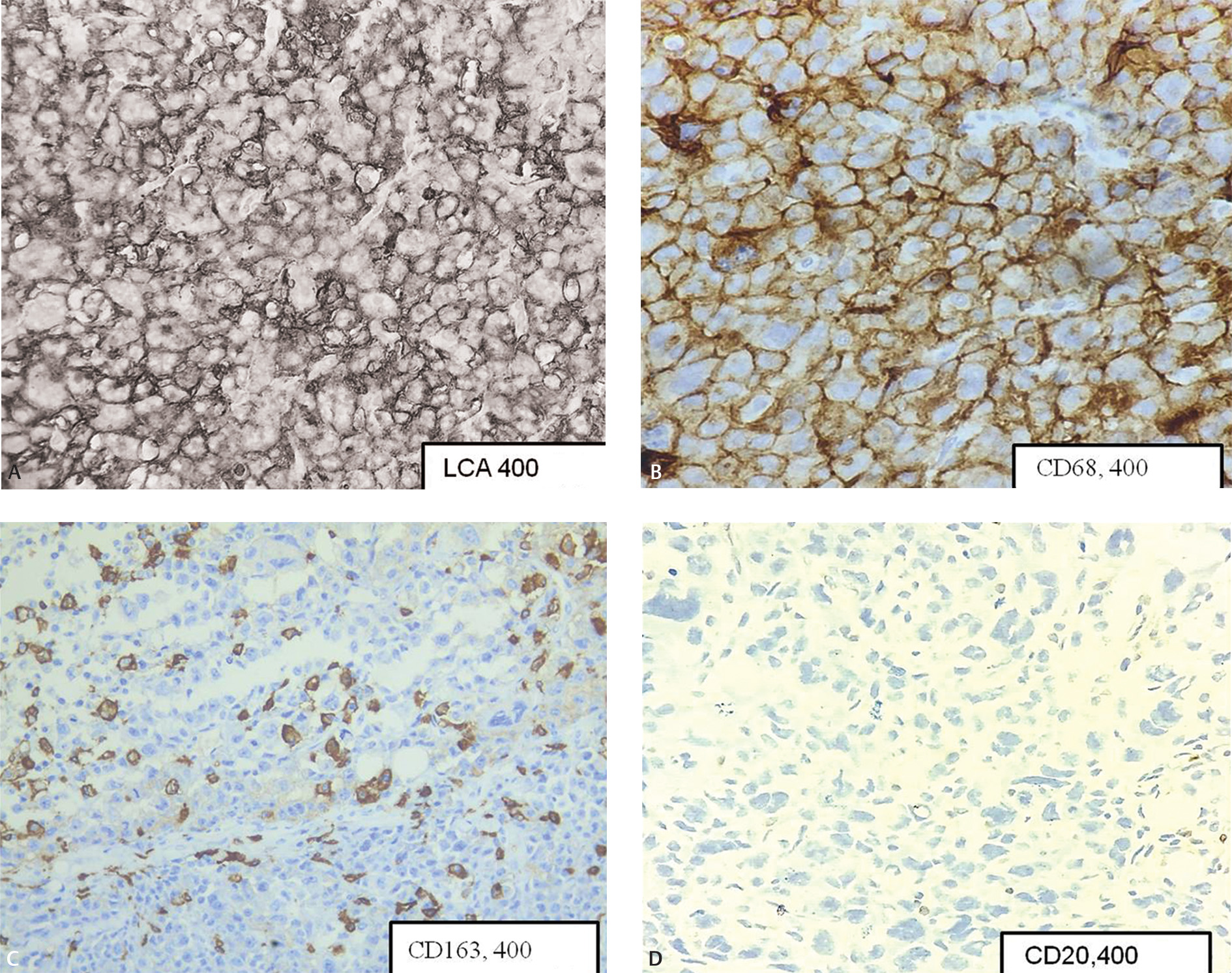
A 66-year-old gentleman had presented with generalized lymphadenopathy and was diagnosed elsewhere as diffuse large B cell lymphoma (DLBCL) 6 months back. Patient had opted for chemotherapy at the present hospital. Immunohistochemistry (IHC) done at presentation showed DLBCL of germinal center phenotype (CD20, CD10, and BCL6 positive).The patient was started on R-CHOP (rituximab, vincristine, cyclophosphamide, and doxorubicin). Positron emission tomography-computed tomography (PET-CT), done at 3 months, showed response to the treatment with regression in the size of lesions. At 6 months on therapy, the patient showed worsening of symptoms in the form of increasing breathlessness and myalgia. Blood investigations showed normal hemoglobin and white blood count (WBC) count and mild thrombocytopenia. LDH levels were 1640 U/L (range: 120–246 U/L). PET-CT showed generalized lymphadenopathy and marked increase in the number and size of the lesions. An excisional biopsy was performed from left axillary lymph node. Histopathological examination showed lesion arranged in sheets.The cells were very pleomorphic, showing large vesicular nuclei, prominent nucleoli, and abundant eosinophilic cytoplasm (Fig. 1AB). Bizarre mitotic figures were present. Few tumor giant cells were also seen. Areas of necrosis were present. IHC was positive for leucocyte common antigen (LCA), CD68, CD163 (Fig. 2AC) S100, CD4, and negative for (pancytokeratin) PCK, HMB45, CD20 (Fig. 2D), CD79a, PAX5, CD10, CD3, CD30, CD138, ALK1, CD34, myeloperoxidase, CD1a, CD21, and BCL2. With the positive reaction for LCA and histiocytic markers, CD163 and CD68 diagnosis of histiocytic sarcoma (HS) developing secondary to DLBCL was rendered. The patient had persistent neutropenia and succumbed within 20 days of diagnosis due to sepsis.
-
Fig. 1 (A) Section shows cellular lesion with large area of necrosis (H&E, ×100). (B) Section shows large pleomorphic cells. Atypical mitotic forms seen (H&E, ×400). H&E, hematoxylin and eosin.
-
Fig. 2 (A) Diffuse membrane staining of LCA (×400). (B) Diffuse staining of CD68 (×400). (C) Cytoplasmic staining in neoplastic cells (×400). (D) Neoplastic cells are negative for CD20 (×400).
Discussion
HS is a rare hematolymphoid neoplasm arising from histiocytic/dendritic cell lineage.1 The incidence of primary histiocytic sarcoma is <1%.2 It is known to arise in association with mediastinal germ cell tumors and (NHL), the occurrence is very rare. Common associations described in literature are follicular lymphoma, small cell lymphoma, DLBCL, splenic marginal zone lymphoma, mantle cell lymphoma, and lymphoblastic leukemia/lymphoma.2 Occurrence of HS with any of these entities might be synchronous or may develop at a later stage of disease. The time duration described in literature is much variable (range: 1–312 months).2
In the present case, the tumor developed at 6 months of therapy, showed an undifferentiated morphology, comprising of pleomorphic bizarre cells. Differential diagnosis considered were recurrence of DLBCL, with features of anaplasia. Loss of CD20 can occur postrituximab, however, absence of other markers like CD10, CD79a, and Pax5 cannot be explained. Carcinoma and amelanotic melanoma were also considered and negated by use of an extensive panel of IHC. Expression of markers like CD68,CD4, and S100 hinted toward the histiocytic nature of the lesion. CD163 is considered to be a specific histiocyte marker was positive in fraction of cells.2 Most of the reviewed publications have described complete loss of B cell antigens except presence of Pax5 in two cases by Shao et al3 in HS developing post-NHL. Also, HS developing from DLBCL is described most commonly seen in low-grade lymphoma like follicular and CLL/SLL. Only one case of HS developing from DLBCL is described by Bassarova et al (Table 1).4 One of the case described by Wang et al,5HS and DLBCL cooccurred in a known case of follicular lymphoma.
|
Author |
Age (y)/sex |
Primary diagnosis |
IHC profile |
Treatment and follow-up |
Diagnosis at relapse |
IHC profile |
Treatment and follow-up |
|---|---|---|---|---|---|---|---|
|
Bassarova et al4 |
62/M |
DLBCL, tongue |
CD20+, CD79a+, PAX5+, CD10+, BCL6+, BCL2+, CD14, CD68, PU1+, CD1a, S100, MAF-B, t(14, 18)+ |
R-CHOP (8 cycles) |
Histiocytic sarcoma, tongue, and skin lesions at 1 year |
CD20, CD79a, PAX5, CD10, BCL6+, BCL2, CD14, CD68+, PU1+, CD1a, S100+, MAF-B- |
MIME chemotherapy, 40 Gy radiation to involved fields, relapsed and succumbed to disease |
|
Present case |
66/M |
DLBCL |
CD20+, CD79a+, PAX5+, CD10+, BCL6+, BCL2+, MUM1, ki67–60% |
R-CHOP (6 cycles) |
Histiocytic, sarcoma, presented, with supra and infradiaphramatic LN, peritoneal, and omental deposits |
CD20, CD79a, PAX5, CD10, BCL6+, BCL2, LCA+, CD68+, CD163+, CD4+, S100+, PCK, HMB45 |
Succumbed to disease |
Abbreviations: DLBCL, diffuse large B cell lymphoma; F, female; HS, histiocytic sarcoma; IHC, immunohistochemistry; LN, lymph node; M, male; MIME, methyl-GAG, ifosfamide, methotrexate, etoposide; R-CHOP, rituximab, vincristine, cyclophosphamide, doxorubicin.
Various mechanisms have been postulated with regard to the change in lineage occurring in NHL (mature lymphoid cell) to HS (which arises from myeloid progenitor cells). The most popular being direct transdifferentiation, wherein lineage switch occurs with inactivation of transcription factors Pax5 which is responsible for terminal B cell phenotype and upregulation of markers of myeloid differentiation via PU1 and CEBPα, CEBPβ3, or occurrence of epigenetic changes in the neoplastic cells which act as switch between the lymphoid and myeloid differentiation.2 Other possible mechanism described is dedifferentiation to a common precursor phenotype which then redifferentiates to HS.2
Fluorescent in situ hybridization and molecular studies could not be done due to financial constraints which is a drawback in the present study. Presence of clonally similar mutations at presentation and recurrence could establish the link between this lineage switch.
Stoecker and Wang6 have postulated that change in lineage does not affect genotype of the tumor. The clonal nature of which can be proven by use of immunoglobulin gene rearrangement studies which can demonstrate same size of amplicons or persistence of the initial cytogenetic abnormality. Feldman et al7 were the first to demonstrate persistence of t(14,18)/BCL2 in eight cases of HS developing from follicular lymphoma.7 Shao et al have also shown persisting clonal genotype in two of the seven cases of Interdigitating dendritic cell tumor/HS developing from CLL/SLL.5
The prognosis is bad as HS are known to be very aggressive neoplasms. In a study by Stoecker and Wang the median survival was 6.5 months (available in 4 of the 22 cases studied).
The case is presented to highlight the challenges faced when encountering a case of second malignancy. An extensive panel of IHC is needed to reach the diagnosis. Molecular studies and sequencing are important for diagnosis and are still elusive in resource poor settings. To our knowledge, this is the first case of HS developing from DLBCL to be reported from India.
Conflict of Interest
None declared.
References
- Histiocytic sarcoma. In: Swerdlow SH, Campo E, Harris NL, eds. WHO classification of tumours of haematopoietic and lymphoid tissues (4th ed.). Lyon, France: IARC Press; 2017. p. :468-469. Revised
- [Google Scholar]
- Histiocytic sarcoma as a secondary malignancy: pathobiology, diagnosis, and treatment. Eur J Haematol. 2016;97(01):9-16.
- [Google Scholar]
- Clonally related histiocytic/dendritic cell sarcoma and chronic lymphocytic leukemia/small lymphocytic lymphoma: a study of seven cases. Mod Pathol. 2011;24(11):1421-1432.
- [Google Scholar]
- Transformation of B cell lymphoma to histiocytic sarcoma: somatic mutations of PAX-5 gene with loss of expression cannot explain transdifferentiation. J Hematop. 2009;2(03):135-141.
- [Google Scholar]
- Sequential development of histiocytic sarcoma and diffuse large b-cell lymphoma in a patient with a remote history of follicular lymphoma with genotypic evidence of a clonal relationship: a divergent (bilineal) neoplastic transformation of an indolent B-cell lymphoma in a single individual. Am J Surg Pathol. 2011;35(03):457-463.
- [Google Scholar]
- Histiocytic/dendritic cell transformation of B-cell neoplasms: pathologic evidence of lineage conversion in differentiated hematolymphoid malignancies. Arch Pathol Lab Med. 2013;137(06):865-870.
- [Google Scholar]
- Clonally related follicular lymphomas and histiocytic/dendritic cell sarcomas: evidence for transdifferentiation of the follicular lymphoma clone. Blood. 2008;111(12):5433-5439.
- [Google Scholar]




