

Colonic adenocarcinoma presenting as hemophagocytic syndrome
Address for correspondence: Dr. Arvind Kumar, Department of Medicine, 3097, Teaching Block, All India Institute of Medical Sciences, New Delhi - 110 029, India. linktoarvind@gmail.com
This article was originally published by Thieme Medical and Scientific Publishers Private Ltd. and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Hemophagocytic syndrome (hemophagocytic lymphohistiocytosis [HLH]) is a rare and potentially fatal disorder characterized by pathological immune activation associated with primary familial disorder, genetic mutation or occurring as a sporadic condition. The later can be secondary to infections, malignancies, or autoimmune diseases. Malignancy-associated HLH is commonly seen in hematological malignancies and rarely with solid organ tumors. We report a case of adenocarcinoma colon presenting as hemophagocytic syndrome. To the best of our knowledge, it is the first case report of HLH secondary to carcinoma colon.
Keywords
Adenocarcinoma colon
hemophagocytic lymphohistiocytosis
hypertriglyceridemia

Introduction
Hemophagocytic syndrome is a systemic hyperinflammatory disease induced by pathologic immune activation, with proliferation of well-differentiated macrophages/histiocytes and increased phagocytosis of the affected organs. The syndrome was first described in 1939 by Scot and Rob and again in 1952 by Farquhar and Claireaux.1 In recent years, hemophagocytic lymphohistiocytosis (HLH) has attracted growing attention due to an apparent inexplicable rise in its occurrence. The incidence of HLH in pediatrics is estimated as 1–225/300,000 per live births and has wide geographic variations.2 Pediatric HLH typically presents in the first year of life with unremitting fever, hepatorenal, and bone marrow dysfunction with or without a positive family history. It is a recessive condition caused by mutations in genes important for natural killer (NK) and T-cell cytotoxic function. Homozygous null mutations in several genes have been implicated in primary HLH that causes familial HLH, FHL2–FHL5; these include PRF1, UNC13D, STX11, and STXBP2.3 Patients with congenital immune deficiency syndrome can also develop HLH due to mutations in gene that regulate immune function, for example, Chediak-Higashi syndrome, Hermansky-Pudlak syndrome, etc.4 Secondary HLH is mostly age independent and it is more common in adults, older children compared to young children. No large epidemiological data is available on secondary HLH on this date. HLH in adults is a heterogeneous entity occurring in the setting of various infectious, malignant, rheumatologic, or metabolic conditions with varied clinical manifestations. Due to rarity of disease, much of our current understanding of pathology and management of HLH has been derived from research in pediatric population with primary HLH. This article presents a case of carcinoma colon in an adult presenting as HLH.
Case Report
A 42-year-old male, resident of Jammu, a mechanic by occupation, an ex-smoker (SI-20) and occasional alcoholic, with no prior comorbidities, presented with intermittent mild to moderate grade evening rise of temperature for the 1 year with increased intensity for the 25 days before admission. Constitutional symptoms were present in the form of loss of appetite and significant loss of weight (10 kg over 2 months). He was initially evaluated in a small private hospital and was found to have pancytopenia with relative lymphocytosis with ultrasound abdomen showing hepatosplenomegaly and enlarged retroperitoneal lymph nodes. Monteux test was positive (15 mm), for which he was started on 4-drug antitubercular treatment (ATT) (duration of ATT intake at admission: 21 days), but there was no symptomatic improvement. He was admitted at our center with these complaints for evaluation.
General physical examination was unremarkable except for the presence of severe pallor. Mild hepatosplenomegaly was present with a soft to firm liver palpable 3 cm below the subcostal margin in midclavicular line and a soft to firm spleen palpable 2 cm below the subcostal margin along the splenic axis. Rest of the systemic examination was unremarkable.
The differential diagnoses at admission were lymphoreticular malignancy, tuberculosis, kala-azar, and brucellosis. The patient was started on treatment as per febrile neutropenia protocol (absolute neutrophil count at admission: 330). Brucella, Epstein–Barr virus, kala-azar (rk39), and enteric serology (widal) were found to be negative, and peripheral smear was negative for malarial parasites. Erythrocyte sedimentation rate and C-reactive protein were highly elevated (132 mm and 96 mg/L), and serum ferritin at admission was very high (4205, with upper limit of normal being 400). Serum fibrinogen levels were normal. The patient's counts continued to fall, requiring a cumulative transfusion of four units of packed red blood cells, seven units of random donor platelets, and four units of fresh frozen plasma. The provisional diagnosis was revised to HLH (secondary to lymphoma) (5 of 8 criteria were met initially: Fever, cytopenias, splenomegaly, elevated ferritin, and bone marrow aspirate showing hemophagocytosis and, later, NK cell activity was also found to be very low, satisfying the 6th criteria) and he was started on dexamethasone at a dose of 10 mg/m2 with septran and fluconazole as per HLH 2004 protocol. Etoposide and cyclosporine were not started as he was clinically stable and these drugs would have interfered with the management of unknown primary tumor. He became afebrile, and his counts progressively rose. Ultrasonography (USG)-guided lymph node biopsy Figure 1 done from mesenteric lymph nodes showed features suggestive of an adenocarcinoma with no evidence of a lymphoma. Contrast-enhanced computed tomography (CT) abdomen Figure 2 revealed multiple enlarged heterogeneously enhancing lymph nodes in the mesentery, periportal region, and retroperitoneum, with area of necrosis. In addition, there was nodular enhancing wall thickening in the region of proximal descending colon with surrounding fat streakiness. However, there was no proximal bowel dilatation. No focal liver or adrenal lesions were seen. There was mild hepatosplenomegaly and no ascites. Imaging suggested the possibility of colonic malignancy with metastatic lymphadenopathy; however, latter was far more extensive and also in nondraining areas of the colon. Colonoscopy suggested a polypoidal growth in the ascending colon from which biopsy was taken. Whole body positron emission tomography-CT Figure 3 showed metabolically active metastatic disease involving mesenteric and retroperitoneal lymph nodes with soft tissue thickening of descending colon with tracer uptake suspicious of primary. Biopsy from the colonic growth Figure 4 showed adenocarcinoma. The final diagnosis was thus kept as HLH secondary to adenocarcinoma colon (stage IIIc-T3N2bM0).
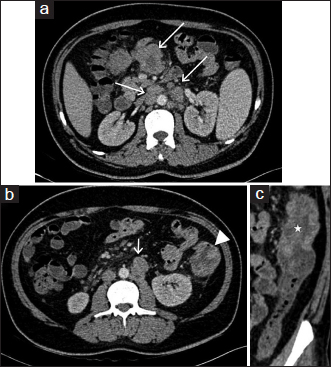
- Axial contrast-enhanced computed tomography images (a and b) show multiple enlarged heterogeneously enhancing lymph nodes in mesentery and retroperitoneum (arrows). (b) Depicts nodular wall thickening in the region of proximal descending colon (arrowhead). (c) Oblique coronal multiplanar reformat image better shows the nodular wall thickening in the proximal descending colon (asterisk). Note is made of mild pericolonic fat stranding
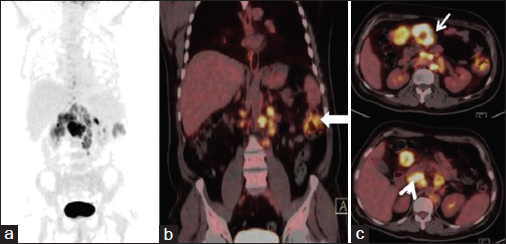
- (a) Maximum intensity projection image (b) coronal fused positron emission tomography-computed tomography and (c) transaxial fused positron emission tomography-computed tomography images reveal abnormal tracer accumulation in the descending colon (thick arrow) and multiple enlarged nodes in the mesentry (thin arrow) and peripancreatic (arrowhead) locations showing avid 18F 2-fluoro-2-deoxy-d-glucose uptake
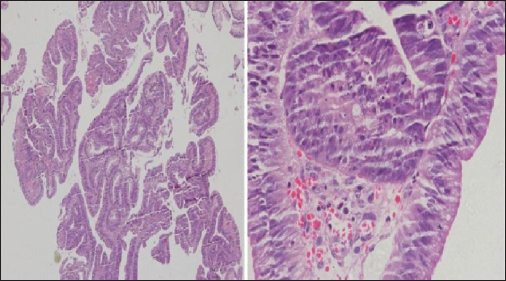
- Colonic biopsy
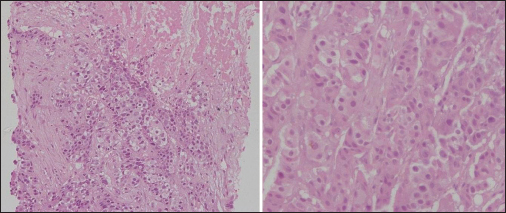
- Mesenteric lymph node biopsy
In view of advanced stage carcinoma colon with florid lymph node metastasis to even nondraining sites and in the absence of any obstructive symptoms, the patient was not taken up for surgery and chemotherapy with FOLFOX (Folinic acid, Fluorouracil, Oxaliplatin-based)-6 (q2wk) regimen was started. While tapering doses of dexamethasone as per HLH protocol, patient again developed pancytopenia. Dexamethasone dose was increased to initial level along with administration of first cycle of chemotherapy. The patient has so far received two cycles of chemotherapy with a modest improvement in cell counts. Serum ferritin came down to 428.
Discussion
HLH is a rare hyperinflammatory disease with dismal prognosis if not promptly treated.5 Traditionally, disease has been divided into primary and acquired. Primary HLH is caused by specific mutations in genes effecting cytolytic activity of T-lymphocytes and NK cells.6 Secondary HLH is associated with an inciting factor that includes infectious, malignant, or rheumatologic conditions. Malignancy-associated HLH is most commonly seen with lymphoreticular malignancies7 but, in recent years, HLH has been reported in association with solid organ tumors of lungs, prostate, and stomach.8 To the best of our knowledge, it is the first case of HLH-associated carcinoma colon to be reported in literature. Oliveria et al. reported a case of HLH in a patient operated for carcinoma colon. Postoperatively, patient developed peritonitis, HLH was attributed to that rather than the malignancy.9
High clinical suspicion is of utmost importance for the diagnosis and early treatment. Diagnosis is suspected from clinical, and laboratory criteria proposed by histiocyte society are made in the presence of at least 5 of 8 criteria10 Table 1. Utility of these criteria is questionable because of lack of specificity. However, some authors argue that despite the lack of specificity of individual criteria that reflects the disease, severity is crucial point. Fever and spleen enlargement are present in 75% of patient. Bicytopenia, hypertriglyceridemia, and serum ferritin level >500 μg/l are present in 50% of cases. Literature review suggests that ferritin level >10,000 μg/l is highly sensitive and specific and at levels >30,000 μg/l, the specificity for HLH reaches 100%.11
|
Number |
Criteria |
|---|---|
|
1 |
Fever |
|
2 |
Splenomegaly |
|
3 |
Cytopenias affecting two or more lineages HB <9 g/dl Platelets <100,000/cumm Neutrophils <1000/cumm |
|
4 |
Hypertriglyceridemia and/or hypofibrinogenemia TG >265 mg/dl Fibrinogen <150 mg/dl |
|
5 |
Serum ferritin >500 µg/l |
|
6 |
Hemophagocytosis in bone marrow, spleen, or lymph nodes |
|
7 |
Decreased NK cell activity |
|
8 |
Soluble IL-2 receptor >2400 U/ml |
HLH diagnosis requires a molecular diagnosis consistent with HLH or 5/8 above criteria. HLH - Hemophagocytic lymphohistiocytosis; Hb - Hemoglobin; TG - Triglyceride; NK - Natural killer; IL-2 - Interleukin-2
In this case, the initial differential diagnosis was lymphoreticular pancytopenia and USG documented mesenteric and retroperitoneal lymph nodes. Negative workup for chronic endemic infections and investigation revealing high serum ferritin, almost up to the level specific for HLH, raised the suspicion of hemophagocytic syndrome, with bone marrow aspirate showing some evidence of hemophagocytosis. Treatment for HLH was started with report of NK cell activity awaited (later came as very low).
The front line treatment for primary HLH is the HLH-2004 protocol. This consists of an 8 weeks induction therapy with dexamethasone, etoposide, and cyclosporine A. There are no prospective studies to guide treatment in adults with secondary HLH. Most experts suggest treatment of primary condition in mild cases. Severe cases must be treated as per HLH-2004 protocol along with the treatment of underlying condition.12 In this case, dexamethasone monotherapy was started for two reasons. First, HLH, in this case, was mild with no evidence of organ dysfunction except for pancytopenia and, second, rapidly falling blood counts precluded the use of etoposide for the fear of bone marrow suppression, thus further decreasing the blood cell counts. Treatment of malignancy-associated HLH is complicated and must be individualized depending on type and stage of tumor, severity of HLH, and tolerability of patient for chemotherapy.
Conclusion
HLH is a rare entity that needs a high index of clinical suspicion for diagnosis and must be suspected in any patient presenting with unremitting fever with pancytopenia. A thorough evaluation for inciting factor including solid organ tumor should be carried out in all adult patients with suspected HLH. Treatment of HLH in the given clinical scenario may be very challenging for practicing physicians, and an individualized approach is the need of the hour.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science. 1999;3(286):1957-9.
- [Google Scholar]
- Girls homozygous for kinase mutation that leads to protein deficiency develop fatal HLH. J Clin Invest. 2009;119:1350-5.
- [Google Scholar]
- How I treat hemophagocytic lymphohistiocytosis in the adult patient. Blood. 2015;125:2908-14.
- [Google Scholar]
- A prospective evaluation of degranulation assays in the rapid diagnosis of familial hemophagocytic syndromes. Blood. 2012;119:2754-63.
- [Google Scholar]
- Approach to hemophagocytic syndromes. Hematology Am Soc Hematol Educ Program. 2011;2011:178-83.
- [Google Scholar]
- The hemophagocytic syndrome in prostate cancer revealed by disseminated carcinomatosis of the bone marrow. J Urol. 2002;168:1101-2.
- [Google Scholar]
- HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124-31.
- [Google Scholar]
- Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002;100:2367-73. et al.
- [Google Scholar]




