

Translate this page into:
Myeloid sarcoma: A 7-year retrospective study from a tertiary cancer care center, illustrating the clinical, morphological, and immunohistochemical features
 MD, PDCR, Sandhyadevi Gundimeda1PhD, Pavankumar Boyella2MD, DM, ECMO, Veerendra Patil2MD, Krishnamohan Mallavarapu2DNB, DM, Senthil Rajappa2MD, DNB, DM
MD, PDCR, Sandhyadevi Gundimeda1PhD, Pavankumar Boyella2MD, DM, ECMO, Veerendra Patil2MD, Krishnamohan Mallavarapu2DNB, DM, Senthil Rajappa2MD, DNB, DM
Corresponding author: Manasi Chetan Mundada, Department of Pathology, Basavatarakam Indo American Cancer Hospital and Research Institute, Hyderabad, Telangana, India. manasicmundada@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Mundada MC, Ahmed F, Kodandapani S, G S, Boyella P, Patil V, et al. Myeloid sarcoma: A seven year retrospective study from tertiary cancer care center, illustrating the clinical, morphological and immunohistochemical features. Asian J Oncol. 2024;10:12. doi: 10.25259/ASJO_35_2023
Abstract
Objectives
Myeloid sarcoma (MS) is a tumor mass formed by the collection of myeloid blasts at sites other than bone marrow. MS can occur as an isolated mass or concurrently as acute myeloid leukemia (AML) in bone marrow or in a case of myeloproliferative neoplasm/myelodysplastic neoplasm. The aim of the study was to highlight the varied presentation and morpho-immunohistochemistry guide to recognize this entity in biopsy for its therapeutic connotation.
Material and Methods
The present study is a retrospective analysis; cases diagnosed as MS between 2014 and 2021 were included in the study. Clinicopathological details like age, gender, site of biopsy, bone marrow status, histopathology, immunohistochemistry, treatment, and follow-up information, where available, were included.
Results
A total of 24 cases were diagnosed with the age range of 2–67 years (Mean 32.08 ± 17.38 years), male: female ratio was 1.1:1. The sites at presentation were: spinal/paraspinal region, lymph nodes, breast, cervix, orbit, nasal cavity/nasopharynx, skin, mediastinum, cheek swelling, central nervous system, and chest wall. De novo MS was present in seven cases (29%), while concurrent AML in peripheral blood /bone marrow was noted in six cases (25%). Histopathological analysis revealed three morphological types: differentiated, monocytic, and undifferentiated. IHC done in 22 cases showed positive for Leucocyte common antigen (LCA) (14/18), MPO (16/22), CD117 (20/22), CD34 (12/22), CD56 (5/10), and CD68 (4/11). Molecular markers that were positive were RUNX1::RUNX1T1, NPM1, and BCR::ABL1 in three, two, and two cases, respectively.
Conclusion
The present case series showcases the common and uncommon clinical presentation of MS cases. The varied sites of involvement, non-diagnostic radiological features, and undifferentiated morphology make diagnosis a challenge. The authors suggest in tandem use of broad IHC panel, flow cytometry, and molecular studies for arriving at the right diagnosis.
Keywords
Myeloid sarcoma
Morphology
Immunohistochemistry
Flow cytometry
Molecular profile

INTRODUCTION
Myeloid sarcoma (MS) is a tumor mass formed by the collection of myeloid blasts at sites other than bone marrow.[1] These masses can be single or multiple in number and show variable maturation patterns: granulocytic, monocytic, undifferentiated, or rarely promyelocytic.[1,2] Historical perspective shows that this lesion has undergone many renaming processes. It was initially described by burns in 1811[3] followed by king in 1853,[4] who very elaborately described it in the autopsy findings of a 7-year-old girl. From the green color imparted by the tumorous masses, he coined the name “chloroma”. In 1966, Rappaport renamed this lesion as granulocytic sarcoma, as not all the tumors showed green coloration. The name MS has been used since 2002, when it was coined by world health organisation (WHO).[2] MS can occur as an isolated mass or concurrently as acute myeloid leukemia (AML) in bone marrow or in a case of myeloproliferative neoplasm/myelodysplastic neoplasm.
The incidence of MS presenting de novo and with AML is 1% and 2–9%, respectively.[1] These neoplasms show a small round cell morphology, undifferentiated at times. Hence, these should be differentiated from other round cell tumors like Ewing’s sarcoma, lymphoblastic lymphoma, neuroblastoma, rhabdomyosarcoma, and germ cell tumor in the pediatric population. Small cell neuroendocrine carcinoma, non-hogdkin’s lymphoma, and plasmacytoma form differentials in adults. Immunohistochemistry forms the main modality for correct diagnosis. A broad panel of immunohistochemistry markers with knowledge of confounding markers can help in prudent diagnosis.
The aim of the study was to highlight the varied presentation and morpho-immunohistochemistry guide to recognize this entity in biopsy for its therapeutic connotation.
MATERIAL AND METHODS
The present study is a retrospective analysis; cases diagnosed as MS between 2014 and 2021 were included in the study. The slides were retrieved from the archives, and clinicopathological details like age, gender, site of biopsy, bone marrow status, and treatment and follow-up information where available were included. Histopathological examination(HPE) with hematoxylin and eosin (H&E) stained slides was done with relevant immunohistochemistry (IHC) panel. A panel of markers done included B cell markers: CD20 (L26, Dako), PAX5 (BC/24, cell marque); T cell markers: CD3 (MRQ-39, cell marque); markers for myeloid differentiation: CD117 (YR145, cell marque), myeloperoxidase (MPO) (Polyclonal MPO, Dako); markers suggesting immature/blast phenotype: CD34 (QBEnd10, Dako), TdT (polyclonal TdT, cell marque), CD56 (MR-42, cell marque) positive in neuroblastoma, small cell carcinoma and aberrant expression in acute myeloid leukemia (AML), pancytokeratin (PCK) (AE1/AE3, Dako) positive in carcinoma/tumor of epithelial origin, LCA/CD45 (2B11+PD7/26, Dako) to prove hematopoietic origin of tumor, Oct3/4 (SEMGC, Biocare) for germ cell tumor, CD138 (B-A38, cell marque) plasma cell neoplasm, CD4 (SP35, cell marque) T cell marker, also used for monocytic differentiation, CD68 (KP1, cell marque) monocyte/macrophage system marker, CD99 (EPR3097Y, cell marque) used in the workup of round cell sarcomas, and desmin (D33, Biocare) used to identify rhabdomyosarcoma. Immunohistochemistry was done, few on semiautomated/completely automated ventana/bond platform using the standard procedures. Molecular workup was done in 10 cases presenting with marrow involvement as per the prognostic panel required for risk stratification of AML. Fluorescence in situ hybridization (FISH), polymerase chain reaction (PCR), karyotyping, and next-generation sequencing (NGS) were used as per the financial resources available [Figure 1]. FISH testing was done for RUNX1::RUNXITI translocation. NPM1, FLT3, and BCR::ABL1 were done by PCR; CEPBA was done by sequencing technology. Karyotyping was done on marrow sample. This unavailability of molecular results in all cases can be a potential source of bias as to the representative nature of the data; moreover, the testing was done on marrow samples only.
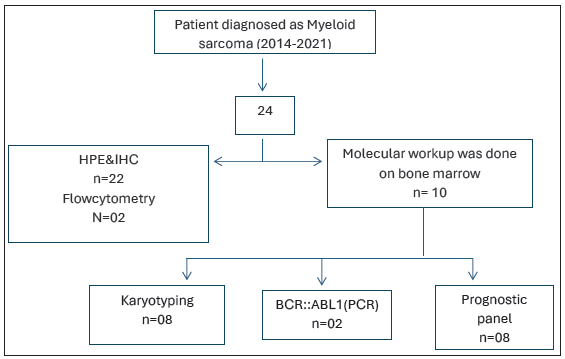
- Flowchart to show workup of the cases. BCR-ABL1: Breakpoint cluster region and abelson murine leukemia 1(name of a gene),PCR: Polymerase chain reaction, HPE: Histopathological examination, IHC: Immunohistochemistry
Statistics: A simple statistical measure percentage was used to express the IHC marker expression patterns.
RESULTS
A total of 24 cases were included in the study. The age range was 2–67 years (Mean 32.08 ± 17.38 years), and the male: female ratio was 1.1:1. The sites at presentation were very varied [Table 1]: spinal/paraspinal region (6), lymph nodes (7), breast (1), cervix (1), orbit (1), nasal cavity/nasopharynx (2), skin (2), mediastinum (1), cheek swelling (1), central nervous system (CNS) (1), chest wall (1). De novo MS was present in seven cases (29%), while concurrent AML in peripheral blood/bone marrow was noted in six cases (25%). Details on bone marrow involvement were not available in six cases. Secondary MS was seen in six cases (25%). Two cases showed concomitant chronic myeloid leukemia, and one case each in chronic phase, blast crisis, and myeloproliferative neoplasm/myelodyaplastic syndrome (MPN/MDS) syndrome favoring juvenile myelomonocytic leukemia (JMML). Of the remaining three cases, one presented as therapy-related MS in treated carcinoma breast, another one presented as CNS relapse in case of acute promyelocytic leukemia (APL), and the third was a case of secondary AML post primary myelofibrosis presenting as relapsed AML. Diagnosis was made on tissue biopsy in all cases except two in which pleural fluid and cerebrospinal fluid (CSF) were the diagnostic samples. The symptoms were also varied according to the site of presentation; spinal lesions were characterized by backache and lower limb weakness, and localized swelling was seen in the nodal, orbital, and oral lesions [Table 1].
| Sr. No | Age | gender | site | Presenting complaints | Presentation | Past history | BMA | t/t |
|---|---|---|---|---|---|---|---|---|
| 1 | 16 | M | D11 lesion | Backache | De novo | - | N | 7+3 |
| 2 | 14 | M | D3 lesion | Not available | I | - | ND | No |
| 3 | 17 | F | Nasopharynx | Not available | I | - | ND | No |
| 4 | 16 | M | Skin | Reddish rashes all over body | S | - | P | 7+3+ASCT |
| 5 | 17 | F | Cervical LN | Painless swelling | S | JMML | P | Aza + ASCT |
| 6 | 9 | F | Cheek swelling | Lower 1/3 face swelling | I | - | ND | No |
| 7 | 2 | M | Retroperitoneal biopsy | Lump abdomen | De novo | - | N | NO |
| 8 | 36 | M | Chest wall | Swelling on chest wall, RVD+ | De novo | - | P | 7+3 |
| 9 | 33 | M | Mediastinum | Sternal swelling | De novo | - | N | NO |
| 10 | 55 | F | Breast | Lump in breast | I | - | ND | No |
| 11 | 19 | M | L4, L5 ED lesion | Weakness in both legs | S | - | P | 7+3 |
| 12 | 27 | F | Inguinal LN | Swelling | De novo | - | N | 7+3 |
| 13 | 65 | M | Paraspinal mass | Swelling | S | k/c/o MS | N | No |
| 14 | 67 | F | Skin/subcutaneous nodule | Multiple swelling over body | S | - | P | No |
| 15 | 44 | F | D5–6 lesions | LL weakness | S | k/c/o ca breast | P | |
| 16 | 23 | M | Inguinal LN | Swelling with skin lesion | S | CML-CP | CML | No |
| 17 | 34 | F | Cervix | Bleeding PV | I | ND | No | |
| 18 | 43 | F | Submental lymph node | Swelling | S | C/O CML-CP | CML | No |
| 19 | 40 | F | Nasal cavity, | Referred as case of fungal sinusitis | I | - | N | No |
| 20 | 38 | M | lymph node | Swelling | S | k/c/o PMF | N | No |
| 21 | 41 | F | Orbit | Orbital swelling | I | - | ND | No |
| 22 | 57 | M | CNS | Headache and numbness in left leg | S | K/C/O APML | N | Arsenic + cranial radiation |
| 23 | 27 | M | Retroperitoneal LN with pleural effusion | K/c/o recurrent pancreatitis, breathlessness | De novo | - | N | 7+3 |
| 24 | 30 | M | Omentum and abdominal lymph nodes | Pain abdomen | S | - | P | No |
I: Indeterminate, S: simultaneous bone marrow involvement, P: positive for involvement, N: negative for involvement, ND: not done, PMF: primary myelofibrosis, APML: Acute promyelocytic leukemia, ASCT: allogenic stem cell transplant, CML: chronic myeloid leukemia, aza-Azacytidine, 7+3-Cytarabine + Anthracycline based induction regimen in AML, M: Male, F: Female, JMML: Juvenile myelomonocytic leukemia, RVD: Retroviral disease, ED: Extradural, MS: Myeloid sarcoma, LL: Lower limb, k/c/o: Known case of, PV: Per vaginum, CML-CP: Chronic myeloid leukemia-Chronic phase, CNS: Central nervous system, BMA: Bone marrow aspiration, t/t: Treatment, LN: Lymph node, APL: Acute promyelocytic leukemia.
Histopathological analysis on H&E staining revealed three morphological types: differentiated, monocytic, and undifferentiated morphologies [Figure 2]. Immunohistochemistry was done in all the cases except two where the diagnosis was established on body fluids. Frequency of the above parameters mentioned in Table 2. Cytogenetics/molecular workup was available in 10 cases; three showed RUNX1::RUNX1T1 fusion by FISH, NPM1 gene mutation was positive in two cases, and BCR::ABL1 was positive in two cases (p210 transcript tested by polymerase chain reaction(PCR)). Cytogenetics done on bone marrow sample showed trisomy 4 in two patients, trisomy 13 in one, translocation t(3;8) in one, and complex karyotype in one patient.
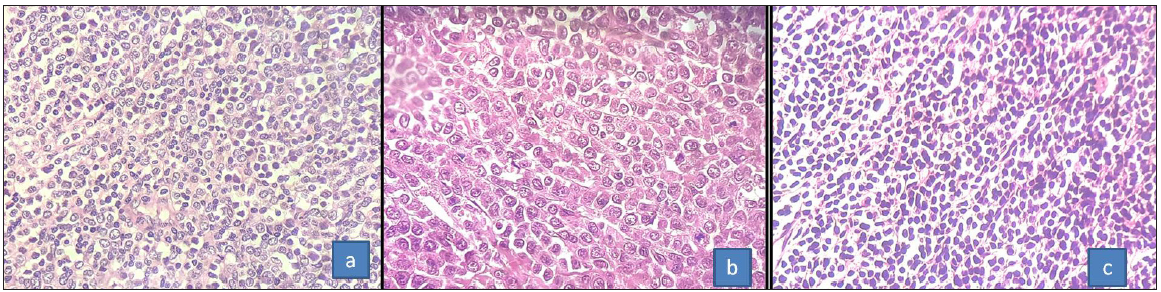
- (a) H&E microphotograph showing the granulocytic morphology with interspersed mature myeloid cells, (b) monocytic morphology, and (c) undifferentiated blastic morphology (400x) H&E: Hematoxylin and eosin.
| Sr.No. | HPE | LCA |
CD 117 |
MPO | CD20 | CD3 | CD68 | CD34 | CD56 | Other IHC | Mol./cytogenetics |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Undiff | ND | P | P | N | N | N | N | ND | 46XY | |
| 2 | Undiff | P | P | P | N | N | ND | P | ND | ||
| 3 | Undiff | ND | P | P | N | N | ND | P | ND | ||
| 4 | Undiff | P | P | N | N | N | ND | N | N | 46XY, ins(5,?), del(7), del(9), del(10), del(12) | |
| 5 | Diff | P | P | P | N | N | P | P | N | 46, XX, t(3;8) (q25;q22) | |
| 6 | Undiff | P | P | P | N | N | N | P | N | t(9,22)-, NPM+ FLT3 & CEBPA- | |
| 7 | Undiff | N | P | N | N | N | N | N | N | Desmin, CD99, PCK-neg | |
| 8 | Undiff | P | P | P | N | N | ND | P | P | t(8,21), FLT3, NPM, CEBPA-Neg. trisomy 4 | |
| 9 | Undiff | P | P | N | N | N | N | P | ND | oct3/4- | |
| 10 | Undiff | ND | P | P | N | N | N | P | ND | ||
| 11 | Undiff | P | P | P | N | N | ND | N | P | t(8,21),Trisomy 4 & 13 | |
| 12 | Undiff | N | P | P | N | N | N | N | ND | ||
| 13 | Undiff | N | P | P | N | N | N | P | ND | ||
| 14 | Mono-cytic | N | N | N | N | N | P | N | P | CD4 + | |
| 15 | Undiff | ND | P | P | N | N | ND | N | ND | PCK- | |
| 16 | Undiff | P | N | P | N | N | ND | P | ND | BCR-ABL+ | |
| 17 | Diff | P | P | P | N | N | P | P | N | PCK,CD99-neg | |
| 18 | Undiff | P | P | P | N | N | ND | P | ND | BCR-ABL+ | |
| 19 | Undiff | P | P | N | N | N | N | N | N | CD138, CD4, CD5-neg | |
| 20 | Undiff | P | P | P | N | P | ND | N | P | ||
| 21 | Undiff | P | P | P | N | N | ND | N | ND | t(8,21), NPM+ | |
| 22 | APL | ND | ND | ND | ND | ND | ND | ND | ND | ||
| 23 |
Mono cytic |
ND | ND | ND | ND | ND | ND | ND | ND | Neg for Myeloid NGS panel | |
| 24 | Undiff | P | P | P | N | N | P | P | P |
Undiff: undifferentiated/blastic, diff: differentiated/granulocytic, P: Positive, N: Negative, ND: not done, PCK: pancytokeratin, IHC: Immunohistochemistry, MS: Myeloid sarcoma, HPE: Histopathological examination, MPO: Myeloperoxidase, LCA: Leucocyte common antigen, NPM1: Nucleophosmin1, FLT3: Fms-like tyrosine kinase 3, EBPA: CCAAT enhancer binding protein alpha, BCR-ABL1: Breakpoint cluster region and abelson murine leukemia 1(name of a gene), NGS: Next generation sequencing.
Six patients received induction chemotherapy with 7 + 3 regimen (Cytarabine infusion for 7 consecutive days with anthracycline infusion on first 3 days of the cycle), one patient was treated with azacytidine, and a case of acute promyelocytic leukemia (APML) was started on arsenic trioxide with radiotherapy.
DISCUSSION
The overall incidence of MS described in literature is around 2–9% of AML cases.[5,6] However, there is a wide variation in the ranges published due to different evaluation criteria used in defining MS; for example, in cases of AML proven on bone marrow/peripheral blood presence of nodal disease many a time does not warrant a biopsy. In the present study, biopsy-proven cases were included along with two cases diagnosed on fluid cytology.
Clinical features
The age range and gender ratio were in concordance with the reported literature.[6,7] The most common site of presentation was lymph nodes and spine. However, rare sites like cervix, nasopharynx, and mediastinum were also noted in this study.
Isolated MS was noted in seven cases in the present study which is a rare finding. Six cases with concomitant AML showed french american british (FAB) M1, M2, and M4 morphologies in marrow. Secondary MS was noted in five cases [Table 1], two of which were MPN (PMF, CML) and one was MPN/MDS (JMML).
Extramedullary proliferation of blasts is a known occurrence in MPN and in transformation to leukemia this needs to be differentiated from MS. MS by definition shows effacement of the architecture of the organ involved by single lineage cells. Another differential diagnosis to be excluded is extramedullary hematopoesis (EMH) seen in cases of MPN.[1,8,9] Though it is more common in hematopoietic organs like spleen and liver, other sites have also been described.[8] Morphologically, proliferation of undifferentiated monomorphic cell population favors the diagnosis of MS over EMH. Also, the presence of cells of multiple lines of differentiation (myeloid/erythroid/megakaryocytic) would point toward EMH. The differentiated type of MS which shows admixed population of precursor blast cells with mature myeloid cells would be a challenging differential diagnosis. IHC can help in resolving this dilemma in some cases. The presence of markers of immaturity CD34, Tdt, and CD117 in the predominant population of cells favors the diagnosis of MS. Also, the presence of aberrant markers like CD19 and CD56 not commonly expressed in normal myeloid cells can be useful to confirm the neoplastic nature of cells. FISH workup for RUNXI::RUNX1T1 and CBFB::MYH11 can also help in these scenarios, as these are commonly associated recurrent genetic abnormalities in MS.
However, there would be a subset of cases, especially de novo MS and those associated with MPN/MDS syndromes which would require a multidisciplinary approach, correlating clinical, hematological, radiological, and at times molecular features in tandem. The presence of MS in MPN is associated with worse prognosis which might be due to the accumulation of various clonal abnormalities leading to complex karyotypes and thereby resistance to treatment.[9,10]
Therapy-associated MS (t-MS) is a rare scenario, most commonly associated with chemotherapy or radiotherapy for solid/hematological malignancies. Carcinoma breast is the most common malignancy in which t-MS is seen and was seen in the present case (Case 15). The patient presented with spinal lesion thought to be metastasis of the epithelial malignancy; however, on HPE and IHC, it was proven to be MS. Further workup showed the presence of bone marrow involvement by AML. JAK2, BCR::ABL1, and KMT2A gene mutations are known to be common in secondary MS.[10] However, BRAF and KMT2A associations are described in t-MS.[11] Molecular workup was not available in the present case.
Pathological features
HPE showed diffuse effacement of architecture of the organ involved with the abnormal cell population. It is interesting to understand what causes the blasts to migrate from the bone marrow into the tissues. Though the exact reason is largely unknown, various theories have been proposed, the principal among them is the altered homing signals to blasts which leave the bone marrow niches and cause extramedullary infiltration.[12,13] This is achieved by an unusual expression of CD56/NCAM1 which is cell adhesion molecule on blasts which dictate migration. CD56 expression is rich in tissues like breast, testis, gut, and ovary which is responsible in homing the blasts in these tissues.[13] Also, deregulation of the CXCR4 expression facilitates accumulation of blasts in visceral organs. Another purported theory involves the role of matrix metalloproteinases (MMP9). High levels of expression of the enhancer of zeste2 (EZH2) attenuates expression of tissue inhibitor of metalloproteinases (TIMP) which in turn upregulates MMP. The uninhibited action of MMP degrades the extracellular matrix and facilitates escape of blasts in extramedullary spaces.[12]
The involved organs showed sheets of neoplastic cells with the presence of residual normal structures in between [Figure 3a]. Lymph nodes showed loss of the follicular pattern, replaced with sheets of cells. There was obliteration of the subcapsular sinuses, and perinodal extension was noted [Figure 3b]. In spinal lesions, in addition to sheets of atypical cells, hyalinization and sclerosis were conspicuous [Figure 3c]. Indian file arrangement was seen in few cases, a finding also noted by Pileri et al.[14] Skin lesions showed predominantly dermal and subcutaneous involvement [Figure 3d]. Periadnexal arrangement was seen in Case 4. Only one of the two cases showed monocytic morphology, an association commonly described in literature.[14]
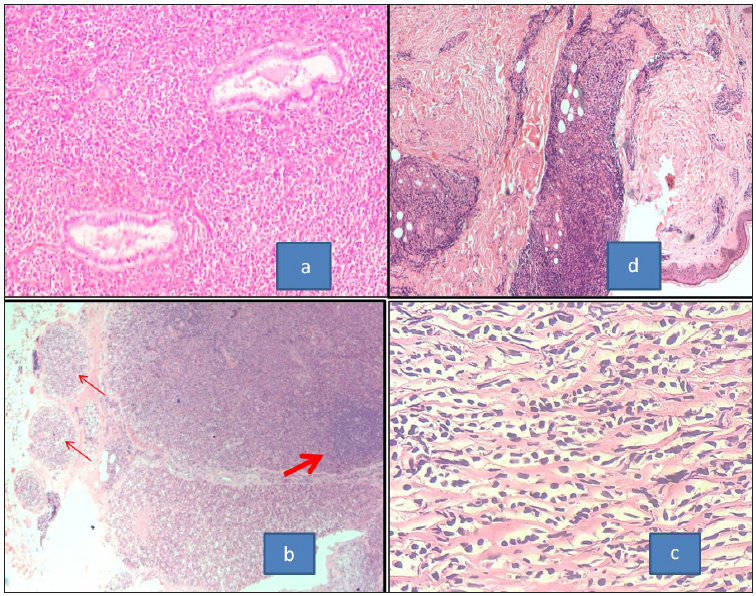
- (a) Section of MS from cervix showing the neoplastic cells going around the normal cervical glands (H& E, 100x). (b) MS involving the lymph node showing effacement of nodal architecture with the peripheral localization of remnant follicles (thick arrow) and presence of neoplastic cells in the afferent vessels (thin arrows) (H&E, 40x). (c) Section from paraspinal mass from case of carcinoma breast showing sclerosis and Indian file arrangement of the neoplastic cells (H&E, 400x). (d) MS involving the skin with the presence of neoplastic cells along dermal vessels and periadnexally (H&E, 40x). MS: Myeloid sarcoma, H&E: Hematoxylin and eosin.
Three morphological subtypes were noted in our series, and the most common morphology was undifferentiated morphology. It comprised monotonous population of medium-sized cells arranged in sheets, showing round nucleus, small inconspicuous nucleoli, and scant cytoplasm. The most common differential diagnosis for this subtype was non-hodgkin lymphoma. In children, round cell tumor, neuroblastoma, rhabdomyosarcoma, and ewing’s sarcoma also formed the differential as seen in Cases 6 and 7. The cells with differentiated patterns showed sheets of medium-sized cells, having round to indented nuclei with eosinophilic cytoplasm. The amount of cytoplasm in this subtype was more and eosinophilic compared to the undifferentiated subtype. Another clue to this subtype was the presence of granulocytes (band forms/neutrophils) and eosinophils admixed with the blastic population. The third subtype was with monocytic morphology, composed of large cells having vesicular nucleus, conspicuous nucleoli, and more amount of eosinophilic cytoplasm. Poorly differentiated carcinoma, large cell lymphoma, and melanoma form the differential diagnosis in this scenario.
IHC was done in 22 cases. The concise panel of IHC was positive for LCA (14/18), MPO (16/22), CD117 (20/22), CD34 (12/22), CD56 (5/10), and CD68 (4/11). All the cases were negative for CD20 except for one, CD3. Some other markers were added to the panel as per case requirements. Pancytokeratin was done to rule out epithelial malignancy. Desmin and CD99 were added to rule out round cell tumor. CD117 was the most sensitive marker positive in 90% [Figure 4]; two cases where it was negative showed monocytic and undifferentiated morphologies. CD117 is a very helpful marker in establishing a myeloid origin of neoplasm with immature phenotype.[8] In monocytic morphology MPO, LCA, and CD117 can be negative; such cases present as a challenge in the diagnosis as hematolymphoid neoplasms. CD43 is a very sensitive marker and hence a useful marker, especially when it is expressed in CD20 and CD3 negative scenarios. In these circumstances, expression of CD43 points toward hematolymphoid malignancy with myeloid lineage differentiation.[7,15] However, CD43 was not used as part of the panel; the absence of CD43 was compensated by use of markers for monocytic differentiation (CD68, CD163, and CD4). Many studies[2,14] have found CD68 to be a very sensitive marker expressed in majority of the cases; our experience has been different with 36% of MS stained positive by this marker. It is a nonspecific marker seen in many hematological neoplasms,[7,8] especially when the KP1 clone is used. In the present study, the PG-M1 clone was used which is more specific for monocytic differentiation,[7,15] hence the lower expression percentage. Also, it was backed by a second monocytic marker in cases with monocytic differentiation. Many authors have also used HLA-DR, CD33, and lysozyme to establish myeloid differentiation. Other nonspecific markers like CD7 and PAX5 are common aberrant cross-lineage antigens that are expressed in AML and can sometimes support the diagnosis. CD56 expression has been postulated to be responsible for homing of the blast cells in extramedullary sites, especially in cases associated with RUNXI::RUNX1T1.[14,16] In the present study, 50% of the cases showed CD56 expression, including two cases of RUNXI::RUNX1T1 which showed CD56 expression. Expression of CD56 in RUNXI::RUNX1T1-positive cases connotes a bad prognosis.[16]
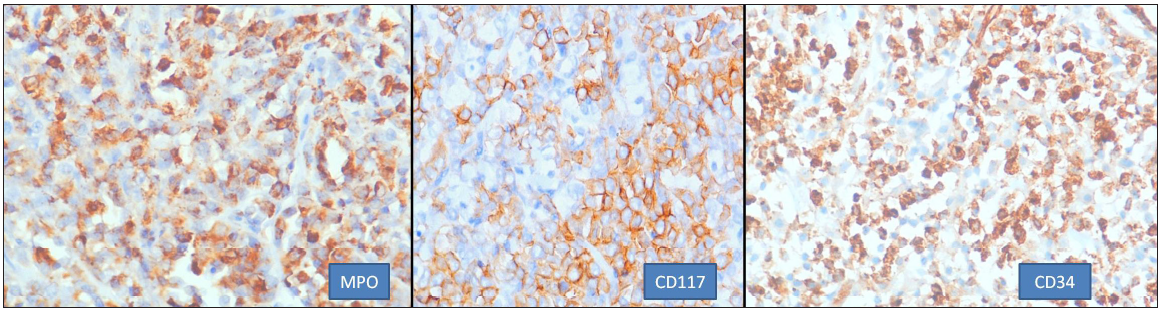
- Common immunohistochemistry markers helpful in diagnosis (400x). MPO: Myeloperoxidase.
In Cases 22 and 23, the diagnosis was done on cerebrospinal fluid (CSF) and pleural fluid, respectively. In both the scenarios, there was risk associated with biopsy procedure, and flow cytometry-based diagnosis alleviated the need for tissue biopsy. Case 22 was a known and treated case of acute promyelocytic leukemia, who presented with symptoms of headache and numbness in the left leg. Relapse with CNS involvement was suspected due to the presence of mass lesion in magnetic resonance imaging (MRI). CSF tapping was done for confirming the disease. The presence of promyelocytes on cytological evaluation confirmed the relapse of disease and the same sample was processed by flow cytometry. However, it failed due to low cell count and cell degeneration. In Case 23, the patient presented with retroperitoneal mass and ongoing acute pancreatitis, due to which biopsy was prohibited. Pleural effusion was present, and tapping was done to relieve respiratory distress. Cyto-morphology of the fluid showed the presence of blasts, which were confirmed of myeloid origin by flow cytometry. Both these scenarios prohibited biopsy and hence confirmatory tissue diagnosis was done by orthogonal modality of testing by cyto-morphology, and flow cytometry was useful. The factors pertinent for using flow cytometry would be the involvement by disease and availability of an ample number of viable cells, since the cells tend to degenerate faster in fluid than the blood sample.
Interesting case scenario
In present study, Case 20 was a diagnosed case of PMF (JAK2+), which progressed to AML. The patient completed induction chemotherapy elsewhere, was documented in remission, and was referred to our center for stem cell transplant. At our center, the patient presented with normal peripheral blood counts, and physical examination revealed a firm cervical lymph node. The node was biopsied. HPE showed involvement of the node by small round cells, and a possibility of MS was raised in lieu of history of AML. In a week’s time, the peripheral blood started to show the presence of blasts. Hence, bone marrow was performed to confirm relapse, which showed the presence of myeloid blasts with small clone (8%) of T lymphoid blasts. This led to reevaluation of the node with IHC, assumed to be MS on morphology. It showed predominant population of T lymphoblasts with admixed myeloid blasts [Figure 5]. Mixed phenotypic blastic transformation is known in MPN.[1,17] This case is presented to emphasize the need for immunophenotyping by IHC in all cases, even those presenting concomitantly with AML.
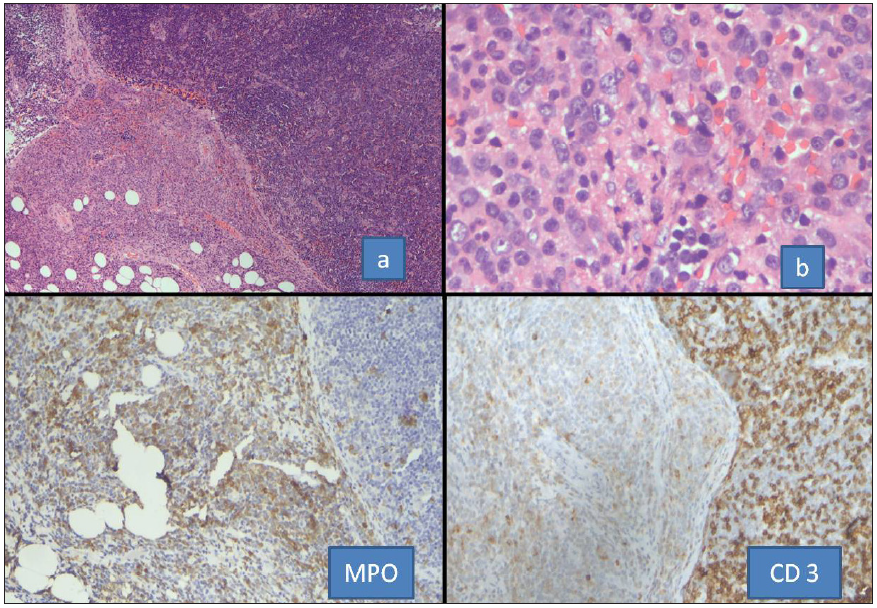
- Sections from the case of JAK2-positive MPN presenting as MS in the lymph node. Sections (a, b) show effacement of node with perinodal spread by neoplastic cells. The extranodal cluster is positive for MPO while the nodal neoplastic cells show CD3 expression. MPO: Myeloperoxidase, MPN: Myeloproliferative neoplasm, MS: Myeloid sarcoma, JAK 2: janus kinase 2.
Molecular workup
The most common molecular abnormalities associated with de novo MS are RUNXI::RUNX1T1, CBFB::MYH11, and KMT2A mutations. The site of MS is shown to have specific molecular associations, for example, orbital MS is known to harbor RUNXI::RUNX1T1, while abdominal MS more commonly show CBFB::MYH11 abnormalities. In our case series, three out of eight cases tested positive for RUNXI::RUNX1T1. The details of cytogenetic abnormalities are shown in Table 2. NPM1 mutation was tested in two cases, and two showed positive results. These mutations are known to present in cases showing monocytic morphology. BCR::ABL1 was positive in two cases.
Treatment
There is no consensus on the treatment of MS; various modalities include chemotherapy, radiotherapy, targeted therapies, and stem cell transplant.[2,18] Chemotherapy is the treatment of choice in predominant cases, especially when there is concomitant involvement of marrow or relapsed MS. Cytarabine- and anthracycline-based chemotherapy regimens as induction akin to those used in AML are used, followed by consolidation with high-dose cytarabine (HiDAC) or allogenic stem cell transplantation. In the present study, eight patients were treated at this center, of which all patients received 7 + 3 AML regimen induction. Six patients received consolidation with HiDAC and two patients in addition underwent allogenic stem cell transplant. Cranial irradiation with arsenic was given in the APML patient who developed central nervous system (CNS) relapse. Radiotherapy is included in the treatment of cases wherein vital structures are compromised, for example, spinal cord compression, superior vena cava obstruction, or as in the present case of isolated CNS involvement. Surgery alone does not have a role in treatment except in a scenario wherein excision biopsies are required to reach a diagnosis.[2] Though at present the role of molecular aberrations in MS is limited in guiding therapy, targeted therapy can be used effectively. Allogenic stem cell transplant is considered in both de novo and concurrent setting, commonly applied post-induction therapy after achieving complete remission.[2]
Limitation of the study
The study setting being a tertiary care center, many cases were received as slides for diagnosis and ancillary testing, and the patients left for treatment at the parent hospital. So, treatment and follow-up details are not available in these cases.
CONCLUSION
The present case series showcases the common and uncommon clinical presentation of MS cases. Several case studies have described the high rate of misdiagnosis in these cases, especially de novo cases of MS. The varied sites of involvement, nondiagnostic radiological features, and undifferentiated morphology make diagnosis a challenge. The authors suggest in tandem use of broad IHC panel, flow cytometry, and molecular studies for arriving at the right diagnosis.
List of abbreviations
AML: Acute myeloid leukemia
APL: Acute promyelocytic leukemia
CNS: Central nervous system
CSF: Cerebrospinal fluid
EMH: Extramedullary hematopoiesis
EZH2: Enhancer of Zeste2
HiDAC: High-dose cytarabine
H & E: Hematoxylin and eosin
HPE: Histopathological examination
IHC: Immunohistochemistry
JMML: Juvenile myelomonocytic leukemia
MMP: Matrix metalloproteinases
MDS: Myelodyaplastic syndrome
MS: Myeloid sarcoma
MPO: Myeloperoxidase
MPN: Myeloproliferative neoplasm
TIMP: Tissue inhibitor of metalloproteinases
t-MS: Therapy-related myeloid sarcoma
Acknowledgements
Authors are grateful for the technical support of Mr Janaiah and Mr K Ramchander Reddy for enabling CBC, flow cytometry, and conventional cytogenetics. They also thank Mrs M Padma Rani for processing blood/bone marrow samples for FISH analysis, and Mr Ravinder and Mr Hussain for the technical help with H&E sections and IHC.
Ethical approval
The research/study approved by the Institutional Review Board at Basavatarakam Indo American cancer hospital and research institute, Hyderabad, Telangana, number IEC/2021/148, dated 29/10/21.
Declaration of patient consent
Patient’s consent is not required as patient’s identity is not disclosed or compromised.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation:
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
References
- Myeloid sarcoma. In: Swerdlow SH, Campo E, Harris NL, eds. WHO classification of tumours of haematopoietic and lymphoid tissues (Revised 4th edn). Lyon IARC Press; 2017. p. :167-68.
- [Google Scholar]
- A case of chloroma. Mon J Med Sci. 1853;17:97. Available from: https://api.semanticscholar.org/CorpusID:51696399 [accessed 2023 Nov 27]
- [Google Scholar]
- Myeloid sarcoma: Current approach and therapeutic options. Ther Adv Hematol. 2011;2:309-16.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Clinicopathological features of myeloid sarcoma: Report of 39 cases and literature review. Pathol Res Pract. 2016;212:817-24.
- [CrossRef] [PubMed] [Google Scholar]
- Benign extramedullary myeloid proliferations. Mod Pathol. 2007;20:405-15.
- [CrossRef] [PubMed] [Google Scholar]
- Extramedullary blastic transformation of primary myelofibrosis in the form of disseminated myeloid sarcoma: A case report and review of the literature. Clin Exp Med. 2020;20:313-20.
- [CrossRef] [PubMed] [Google Scholar]
- Clinicopathological characteristics of de novo and secondary myeloid sarcoma: A monocentric retrospective study. Eur J Haematol. 2018;100:603-12.
- [CrossRef] [PubMed] [Google Scholar]
- Early therapy-related myeloid sarcoma and deletion of 9q22.32 to q31.1. Pediatr Blood Cancer. 2014;61:1701-3.
- [CrossRef] [PubMed] [Google Scholar]
- Myeloid sarcoma: The other side of acute leukemia. In: Guenova M, Balatzenko G, eds. Hematology - Latest research and clinical advances. London, UK: IntechOpen; 2018. doi: 10.5772/intechopen.74931
- [Google Scholar]
- Molecular and cellular aspects of extramedullary manifestations of acute myeloid leukemia. J Cancer Metastasis Treat. 2016;2:44-50.
- [Google Scholar]
- Myeloid sarcoma: Clinico-pathologic, phenotypic and cytogenetic analysis of 92 adult patients. Leukemia. 2007;21:340-50.
- [CrossRef] [PubMed] [Google Scholar]
- Myeloid sarcomas: A histologic, immunohistochemical and cytogenetic study. Diagn Pathol. 2007;2:42.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Multiplex approach in classification, diagnosis and prognostication in acute myeloid leukemia: An experience from tertiary cancer center in South India. Indian J Med Paediatr Oncol. 2017;38:266-72.
- [CrossRef] [PubMed] [Google Scholar]
- Chronic myeloid leukemia, BCR-ABL1-positive. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, eds. WHO classification of tumours of hematopoietic and lymphoid tissues (Revised 4th ed). Lyon: IARC Press; 2017. p. :306.
- [Google Scholar]
- Myeloid sarcoma: Presentation, diagnosis, and treatment. Clin Lymphoma Myeloma Leuk. 2017;17:263-7.
- [CrossRef] [PubMed] [Google Scholar]




