

Translate this page into:
Myelodysplastic syndromes: Where do we stand?
Address for correspondence: Dr. Nitin Sood, Medanta - The Medicity, 10th Floor, Sector 38, Gurgaon, Haryana, India. docnitinsood@yahoo.com
This article was originally published by Thieme Medical and Scientific Publishers Private Ltd. and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Myelodysplastic syndromes (MDS) are fairly common hematological disorder of elderly. They are a group of clonal malignant hematopoietic stem cell disorders characterized by dysplastic morphology, variable cytopenia and a variable threat of transformation to AML. These dysplastic changes are a result of chromosomal abnormalities and somatic mutations. MDS is the most common myeloid neoplasm of the older adults with median age at diagnosis being 72 years and an average incidence rate of 0.2 per 100,000 people per year. MDS is diagnosed and classified according to the WHO 2008 classification system, which utilizes peripheral blood and bone marrow findings. Other essential investigations include flow cytometry, genetic profile and chromosomal analysis. Various prognostic scoring system have been developed which help guide the treatment. Treatment of complications associated with MDS also forms an essential component of the management of this disease.
Keywords
Chemotherapy
hypomethylating agents
myelodysplastic syndromes

Introduction
Myelodysplastic syndromes (MDSs) are group of clonal malignant hematopoietic stem cell disorders best summarized as a paradox of peripheral cytopenia despite a hypercellular bone marrow. The hallmark and distinguishing characteristic of MDS is the abnormal morphology that is observed in immature marrow erythroid, granulocytic and megakaryocytic precursor cells, and also in neoplastic mature blood cells. The clinical significance of these syndromes lies in the variable threat of transformation into acute myeloid leukemia (AML) and various morbidities related to underlying cytopenias such as anemia, bleeding, and risk of life-threating infections. The dysplastic hematopoietic precursors are thought to arise due to chromosomal abnormalities and somatic mutations, which in turn lead to exaggerated apoptosis in the marrow (ineffective hematopoiesis) or clonal evolution to AML. Despite a common pathophysiology, these syndromes have diverse phenotypic presentations ranging from refractory anemia (RA) with minimal risk of transformation into AML to an aggressive subtype with multiple blasts bordering on to myeloid leukemia.
MDS arises from a single transformed primitive multipotent hematopoietic progenitor cell that has acquired multiple mutations resulting in dysplasia and neoplastic transformation. MDS is considered a less-severe end of the spectrum of myelogenous leukemia characterized by lower blast fraction as compared to AML, thus creating a fluid division between them.
MDS is the most common myeloid neoplasm of the older adults with median age at diagnosis of 72 years. As per Surveillance, Epidemiology, and End Results (SEER) statistics review, it is rare before the age of 40 years with the incidence rate of just 0.2/100,000 people per year. Incidence in general population is estimated to be 4.8/100,000 people per year with steadily rising to 29.6/100,000 people per year in those aged between 70 and 79 years of age and increases up to 55.8 in those more than 80 years of age. The incidence has been found to be higher in males as compared to females.1
Majority of cases of MDS are age-related without any particular precipitating factor. However, a minority of cases of MDS have been associated with environmental factors (e.g., exposure to chemicals, benzene, radiation, chemotherapy drugs, or tobacco), genetic abnormalities (e.g., trisomy 21, Bloom syndrome, ataxia telangiectasia, and Fanconi anemia), and other benign hematologic diseases (e.g., paroxysmal nocturnal hemoglobinuria [PNH], congenital neutropenia).2 Familial MDS associated with certain germline mutations has also been reported.
Technological advancements have made it possible to identify recurring chromosomal abnormalities and translocations, which have led to a better understanding into the biology of MDS. These cytogenetic abnormalities identified by karyotype analysis, fluorescence in situ hybridization (FISH), or gene mutation analyses also have prognostic significance and affect treatment planning.
There are three main prognostic systems that have been developed in patients with MDS, which take into account a number of factors such as age, sex, morphologic features, blast percentage, clinical characteristics, the presence of cytopenias, transfusion requirements, and cytogenetic abnormalities. These are International Prognostic Scoring System (IPSS) and a revised IPSS (IPSS-R), World Health Organization (WHO) Prognostic Scoring System (WPSS), and MD Anderson Cancer Center MDS model. Among these, IPSS is most commonly used. Over and above these prognostic scores, various other prognostic indicators have been described such as advanced patient age, presence of comorbidities, poor performance status, total white blood cell count >20,000/cumm, severity of anemia and transfusion dependence, refractory or severe (<30,000/cumm) thrombocytopenia, treatment failure with decitabine, and expression of the Wilms’ tumor gene (WT1), which confer a poorer prognosis.
Clinical Presentation
In view of multiple subtypes, clinical presentation in patients with MDS is varied. These heterogeneous presentations reflect variation in the pathobiology of specific subtypes within MDS and other patient-related characteristics. Presentation can vary from a complete asymptomatic individual detected to have unilineage cytopenia on routine examination while underevaluation for unrelated complaints to a patient with poor Eastern Cooperative Oncology Group (ECOG) performance status in view of life-threatening infections caused due to severe neutropenia or life-threatening hemorrhage related to severe thrombocytopenia. Severe neutropenia and thrombocytopenia at diagnosis are usually associated with advanced disease and a poor prognosis.
Anemia is the most common cytopenia and usually presents as fatigue, weakness, exercise intolerance, or an altered sense of well-being. Less common presentations are infection, easy bruising, or bleeding. Systemic complaints such as fever and weight loss are uncommon and, generally, represent late manifestations of the disease or its complications.
There is no specific physical examination finding associated with MDS. Pallor is present in up to 60% of patients, petechial or purpuric rash in up to 26%, and hepatomegaly and splenomegaly occur in approximately 5% and 10% of patients, respectively.
Acute febrile neutrophilic dermatosis, also known as Sweet syndrome, is also a known presentation in the patients with MDS. In the course of the disease, it often signifies progression and transformation of MDS to myelogenous leukemia. Another cutaneous complication known as myeloid sarcoma or chloroma is now approached as an extramedullary AML.3
Patients with MDS may develop infections as a result of neutropenia and granulocyte dysfunction. The granulocytes are dysfunctional and often result in defective phagocytosis, bactericidal activity, adhesion, and chemotaxis leading to increased susceptibility to bacterial infections. Abnormalities are also noted in adaptive immune system. Bacterial infections are the most common with the skin being the most frequent site. Administration of immunosuppressive agents also often leads to infection caused by other organisms such as fungi, viruses, and mycobacterial infections.4
Patients with MDS are also known to have increased incidence of autoimmune abnormalities and may complicate the disease course. The most common ones are chronic rheumatic heart disease, rheumatoid arthritis, pernicious anemia, psoriasis, and polymyalgia rheumatica.5
Evaluation
In an elderly individual presenting with unexplained cytopenia or monocytosis, a diagnosis of MDS should always be considered. Similar to evaluation for any disease, it begins with clinical history, which should include duration, severity and pattern of cytopenias, infections or bleeding if any in the past and number of blood transfusions received. A history in regards to potential etiological cause should also be elicited including but not limited to nutritional status, medications, HIV infection, alcohol and drug use, occupational exposure to toxic chemicals, and prior treatment with antineoplastic agents or radiotherapy.
In addition to latest blood counts and reticulocyte count, a peripheral blood smear and bone marrow examination are essential. Smears should preferentially be made from freshly obtained blood and specimens exposed to anticoagulants for two or more hours should be avoided. Peripheral smears are essential to determine dysplastic cells, which may be limited to single or may involve all hematopoietic lineages. A posterior-superior iliac crest bone marrow aspirate and trephine (BMAT) is undertaken to evaluate the proportion of hematopoietic cell maturation abnormality, cellularity, presence of ring sideroblasts (RS), and fibrosis. Thrombocytopenia, coagulopathy, or neutropenia should not delay an otherwise doable bone marrow examination. In the event of “dry tap” due to extensive fibrosis, a 2.5 cm trephine with good quality imprint smears are adequate to suggest the diagnosis when subjected to examination by an experienced histopathologist. BMAT samples should be sent for iron staining and FISH for common cytogenetic abnormalities that affect response to therapy such as del (5q), del (7q), +8, MLL (11q23) rearrangements, del (13q), del (12q), del (20q), del (11q), and inv (3). Bone marrow biopsy staining with reticulin is helpful in assessing presence and degree of fibrosis.
As a part of differential work up pretransfusion serum erythropoietin (sEpo) levels, Vitamin B12, red blood cell (RBC) folate levels, serology for HIV infection, and detailed iron studies should be sent. Serum copper and ceruplasmin levels should also be included in initial workup and so should serum zinc levels as both copper deficiency and zinc excess can present with similar peripheral blood picture.
To ascertain the prognosis and guide further treatment via stratifying the disease in accordance to the grading systems, knowledge of bone marrow blast percentage, karyotype/cytogentics (as mentioned above), and cytopenias is necessary. Bone marrow blasts are usually assessed by morphological evaluation and flow cytometry (measured by expression of CD34 on cell surface).
A number of pretreatment tests are needed in the patient of MDS in accordance to the particular line of treatment chosen in a particular individual. In case hematopoietic cell transplantion (HCT) is being considered cytomegalovirus status and full human leukocyte antigen (HLA) typing (A, B, C, DR, and DQ) are required. In low or intermediate-1 risk, MDS patient with normal cytogeneitcs and hypoplastic MDS who are more likely to respond to immunosuppressive therapy (IST) screening for PNH, HLA-DR15 positivity, or STAT-3 mutant cytotoxic T-cell clones is useful.6,7,8,9 Patients with clinical picture suggestive of chronic myelomonocytic leukemia should be tested for 5q31-33 translocations and PDGFR-beta gene rearrangements, and JAK2 mutation analysis should be carried out in those with thrombocytosis.
Last but not the least, performance status of the patient is determined in accordance to ECOG or Karnofsky performance scale.
Diagnosis of Myelodysplastic Syndrome
According to the WHO 2008 classification,10 MDS is characterized by the presence of cytopenia(s), dysplasia in one or more cell lines, ineffective erythropoiesis, and increased risk of development to AML. The morphologic classification of MDS is principally based on blast percentage in peripheral blood and bone marrow, type and degree of dysplasia, and presence of RS as is reproduced in Table 1 below.
|
Disease |
Peripheral blood findings |
Bone marrow findings |
|---|---|---|
|
RCUD; RA; refractory neutropenia; refractory thrombocytopenia |
Unicytopenia or bicytopenia No or rare blasts (<1%) |
Unilineage dysplasia: ≥10% of the cells in one myeloid lineage <5% blasts <15% of erythroid precursors are RS |
|
RA with RS |
Anemia No blasts |
≥15% of erythroid precursors are RS Erythroid dysplasia only <5% blasts |
|
RCMD |
Cytopenia(s) No or rare blasts (<1%) No auer rods <1×109/L monocytes |
Dysplasia in ≥10% of the cells in ≥2 myeloid lineages <5% blasts in marrow No auer rods ±15% RS |
|
RAEB-1 |
Cytopenia (s) <5% blasts No auer rods <1×109/L monocytes |
Unilineage or multilineage dysplasia 5-9% blasts No auer rods |
|
RAEB-2 |
Cytopenia (s) 5-19% blasts Auer rods± <1×109/L monocytes |
Unilineage or multilineage dysplasia 10-19% blasts Auer rods± |
|
MDS-U |
Cytopenia (s) ≤1% blasts |
Unequivocal dysplasia in <10% of cells in one or more myeloid cell lines when accompanied by a cytogenetic abnormality considered as presumptive evidence for a diagnosis of MDS <5% blasts |
|
MDS associated with isolated del (5q) |
Anemia Usually normal or increased platelet count No or rare blasts (<1%) |
Normal or increased megakaryocytes with hypolobated nuclei <5% blasts Isolated del (5q) abnormality No auer rods |
RAEB - Refractory anemia with excess blasts; MDS - Myelodysplastic syndrome; RA - Refractory anemia; RS - Ring sideroblasts; RCMD - Refractory cytopenia with multilineage dysplasia; MDS-U - Myelodysplastic syndrome-unclassified; RCUD - Refractory cytopenias with unilineage dysplasia
Diagnostic workup
The diagnosis of MDS is made after the exclusion of secondary causes of cytopenias/marrow dyspoiesis. The major differential diagnoses are as follows:
-
Vitamin B12/folate deficiency
-
Drug-induced cytopenias (recent cytotoxic therapy)
-
HIV infection
-
Anemia of chronic disorders (infection, inflammation, cancer)
-
Autoimmune cytopenia
-
Chronic liver disease
-
Excessive alcohol intake
-
Exposure to heavy metals
-
Other hematological disorders including acute leukemia (with dysplasia or French-American-British type M7), aplastic anemia, myelofibrosis (in case of MDS with marrow fibrosis), and PNH.
Approach for diagnosis
Patient history and physical examination
-
Clinical history (symptoms of anemia, infections due to neutropenia, tendency for bleeding/bruising due to thrombocytopenia)
-
Family history
-
History of prior chemotherapy and irradiation, occupational exposure, concomitant medication
-
Complete physical examination as spleen size.
Blood tests
-
Total leukocyte count, differential, hemoglobin, platelet count, RBC indices (mean corpuscular volume, mean corpuscular hemoglobin concentration, red cell distribution width), reticulocyte count, NEUT-X, and NEUT-Y
-
Measure RBC-folate/S-folic acid, and serum cobalamins
-
Measurement of homocystein and methylmalonic acid
-
Iron, transferrin (total iron binding capacity), ferritin, lactate dehydrogenase (LDH), bilirubin, haptoglobin, direct antiglobulin test (Coombs test), alanine aminotransferase, aspartate aminotransferase, alkaline phosphatase, albumin, uric acid, creatinine, sEpo, serum protein electrophoresis
-
Screening for HIV, parvovirus B19 (hypoplastic MDS)
-
Screening for hepatitis B and C (in transfusion dependent patients).
Bone marrow aspirate and biopsy
At least 500 cells in bone marrow and 200 cells in peripheral blood should be evaluated in an optimally stained bone marrow and blood smears. For significant dysplasia, dysplastic features should be present in at least in 10% of the nucleated cells in the lineage in consideration.
-
Dyserythropoiesis: Multinuclearity, nuclear fragments, megaloblastoid changes, cytoplasmic abnormalities, RS
-
Dysgranulopoiesis: Nuclear abnormalities including hypolobulation, ring-shaped nuclei, hypo/hypergranulation
-
Dysmegakaryopoiesis: Micromegakaryocytes, large mononuclear forms, multinucleation.
Flow cytometric analysis
Flow cytometry provides a very useful and sensitive tool in assessing dysplasia. The presence of more than two abnormalities has a strong predictive value in indicating MDS. Pattern analysis of the expression of various CD markers in granulocytic, monocytic, and megakaryocytic series provides presumptive evidence of MDS. The presence of more than two abnormalities is a strong indicator of MDS. The range of abnormalities is discussed briefly below.
Granulocytic/monocytic abnormalities
-
Loss of granularity as seen by side scatter compared (SSC) versus CD45 scatter: MDS samples reveal a significantly decreased SSC to controls Figure 1. Flow cytometric analysis is highly sensitive to decreased granularity, although this feature can also be appreciated on morphology
-
Abnormal patterns on CD11b versus CD16 and CD16 versus CD13: Abnormal pattern in these CD markers are commonly encountered in MDS patients. Normally, we see a large population in the CD11b/16 and CD11b/13 double positive region, a pattern which is lost in MDS Figure 2
-
Loss of CD10 on granulocytes: Loss of CD10 on neutrophils is commonly encountered in MDS Figure 3
-
Abnormal CD64 in granulocytes and monocytes: This feature is illustrated by Figure 4
-
Discrete blast population on CD45 versus CD34/117: Presence of discrete blast clusters is a strong pointer toward MDS
-
Nonmyeloid aberrant antigen expression: Expression of nonmyeloid antigens such as CD7/2/56/22 is also a strong pointer toward clonal abnormal myeloid cell population.
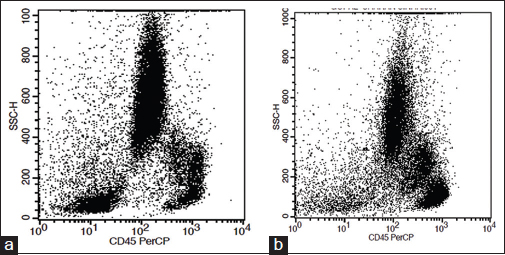
- CD45 versus scatter compared plot. (a) Normal expression pattern. (b) Loss of granularity in myelodysplastic syndrome
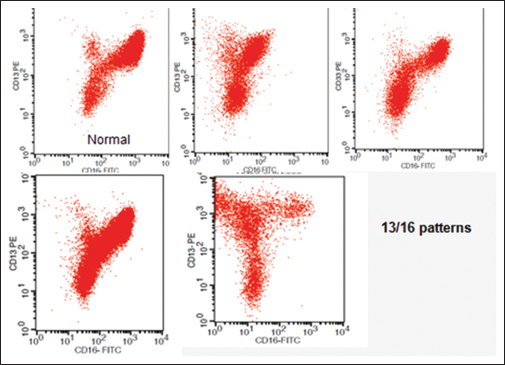
- Abnormal patterns in CD13 versus CD16 plots. Normal controls show an inverted “nike” pattern. Myelodysplastic syndrome graphs reveal different variations compared with normal graphs
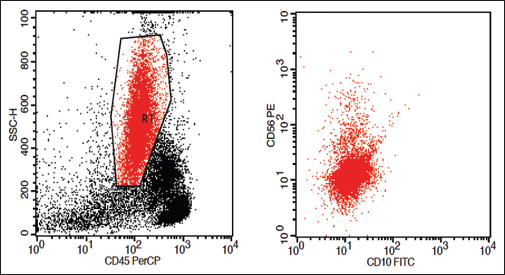
- Loss of CD10 on granulocytes
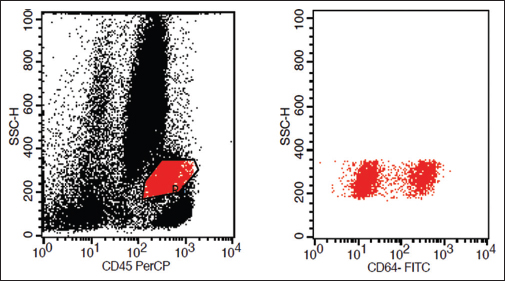
- Loss of CD64 on monocytes
Erythroid abnormalities
Erythroid dysplasia is difficult to assess using flow cytometry due to limited availability of antibodies and challenges in assessing erythroid maturation. Even so, the following patterns may be observed as confirmatory evidence for MDS:
-
Abnormal CD71/glycophorin A (GLYA) patterns
-
Dis-synchronous expression on CD45 versus GLY A plots
-
Dis-synchronous expression on CD45 versus CD71 plots.
Megakaryocytic abnormalities
Megakaryocytic dysplasia is best assessed on morphology at present. This is largely due to limited availability of antibodies as well as limited data on megakaryocytic features in MDS.
Cytogenetic analysis
Cytogenetic abnormalities occur in roughly half of patients with MDS with the most frequent sole abnormalities being del (5q), followed by trisomy 8, -Y, del (20q), and monosomy 7.
Prognosis
Prognosis of patients with MDS can be calculated using a number of established scoring systems. These are as follows:
-
IPSS
-
IPSS-R
-
MD Anderson Comprehensive Scoring System (MDACSS)
-
French Prognostic Scoring System
-
WPSS.
In general, all these scoring systems include analysis of peripheral cytopenias, percentage of blasts in the bone marrow, and cytogenetic characteristics. The most widely adopted scoring system is the IPSS, which was first published in 1997.11
In MDACSS, in addition to the variables in IPSS system, includes patient age, ECOG performance status, and history of prior RBC transfusions.12 WPSS includes recent 2008 MDS classification along with karyotype and severe anemia (<9 g/dL for male and <8 g/dL for female). RA, RA-RS, and MDS with isolated 5q deletion (score 0) have better prognosis than refractory cytopenia with multilineage dysplasia (score 1), RA with excess blasts (RAEB)-1 (score 2) and RAEB-2 (score 3). Even though morphological classification models for MDS are clinically relevant from a prognostic point of view, their relevance in terms of risk stratification is evidently limited in light of the IPSS-R.13
IPSS is being replaced by the new revised score IPSS-R.14 The major differences from the IPSS to the IPSS-R are division into five cytogenetic groups (very good, good, intermediate, poor, and very poor) than three groups in IPSS, dividing patients with <5% blasts into two new groups (<2% and ≥2%), and the magnitude of how low blood cell counts are instead of how many types of cytopenias. This changed the original four IPSS groups to the current five IPSS-R groups: Very low risk, low risk, intermediate risk, high risk, and very high risk.15
In spite of various prognostic scoring systems have been developed and validated for risk stratification in MDS, each of these systems has their limitations which led to recent progress in genomic sequencing techniques that further discoveries of recurrent molecular mutations in MDS patients. These mutations have independent impact on clinical outcomes and overall survival of these patients. Apart from prognostication, this mutational analysis can also help in therapeutic selection for targeted therapy by which a revolution can come in MDS patient care.16
Chromosomal abnormalities
-
Very good: Deletion Y, del (11q)
-
Good prognosis: No chromosomal abnormalities, del (5q), del (20q), del (12p)
-
Intermediate: Del (7q), +8, +19, i17q, other chromosomal abnormalities
-
Poor: Any abnormalities involving chromosome 7, inv (3) having three or more chromosomal abnormalities (“complex”)
-
Very poor: >3 chromosomal abnormalities.
Genetic mutations
Gene expression profiling and chromosomal abnormalities detectable by single nucleotide polymorphism arrays are associated with MDS prognosis.17 After the advent of next generation sequencing, recurrent somatic mutations in genes involved in epigenetic regulation (TET2, ASXL1, EZH2, DNMT3A, IDH1/2), RNA splicing (SF3B1, SRSF2, U2AF1, ZRSR2), DNA damage response (TP53), transcriptional regulation (RUNX1, BCOR, ETV6), and signal transduction (CBL, NRAS, JAK2) have been identified in MDS. Conventional prognostication is by the IPSS-R with additional adverse prognosis conferred by the presence of ASXL1, EZH2, or TP53 mutations.18
The effect of other diseases
A group of Italian researchers developed an MDS-specific comorbidity index that categorizes patients into three risk groups as low (score 0), intermediate (score 1–2), and high (score >2) based on the presence of other comorbidities associated with MDS such as cardiac disease (score 2), moderate to severe hepatic disease (score 1), severe pulmonary disease (score 1), renal disease (score 1), and solid tumors (score 1), which have independent negative effect on prognosis.19
Additional factors effecting prognosis
-
Komrokji et al. showed that the level of the serum protein albumin is an independent factor effecting overall survival in MDS patients, after adjustment for IPSS, age, serum ferritin, and transfusion dependence. Hypoalbuminemia (<3.5 g/dL) associated with low overall survival20
-
Serum LDH, performance status, and ferritin were included in the IPSS-R as supplements to the model.5 In addition, potentially prognostic features include BM fibrosis, beta 2-microglobulin, and flow cytometric profiles. Elevated serum ferritin in the blood resulting from blood cell transfusions and the presence of bone marrow fibrosis are associated with worse prognosis21
-
An additional study concluded that higher numbers of effector memory regulatory T-cells, a type of immune system cell, had a negative effect on survival for lower-risk MDS patients after the researchers controlled for IPSS factors and for the presence of other diseases.
Treatment of Myelodysplastic Syndrome
There is no standard treatment approach for patients with MDS. Historically, the treatment of MDS has been largely unsatisfactory. Currently, only HCT offers a cure. However, multiple new drugs have been approved which can improve symptoms and quality of life, but are not curative. The choice of therapy requires knowledge of the patient's performance status, the patient's age and co-morbidities, the IPSS, IPSS-R risk category, and other disease characteristics.
Not all patients with MDS require treatment. Asymptomatic patients with lower risk MDS may be followed up serially with examinations and laboratory testing to evaluate the disease course. Immediate treatment is indicated for patients with symptomatic anemia, symptomatic thrombocytopenia (e.g., recurrent episodes of bleeding) and patients with recurrent infections due to neutropenia (absolute neutrophil count [ANC] <500).
Supportive therapy, including packed RBCs, platelets transfusion and treatment of infections are the main components of care. In elderly patients with lower risk MDS and multiple co-morbidities, supportive care remains the mainstay of treatment.
Lower risk patients (IPSS low, intermediate-1; IPSS-R very low, low and intermediate; or WPSS very low, low, and intermediate) are primarily treated with supportive care or low-intensity therapies, such as azacitidine, decitabine, lenalidomide or IST. Symptomatic anemia patients with sEpo levels ≤500 mU/mL should be treated with erythropoietin with or without granulocyte-colony stimulating factor. Patients with sEpo levels more than 500 mU/mL, who have a good probability of responding to IST, a trial of antithymocyte globulin (ATG) and cyclosporine, can be given. The most appropriate candidates for IST include younger (<60-year-old) patients, those who are HLA-DR15 positive, those with a hypoplastic bone marrow, those who have a PNH clone, or those who have ≤5% marrow blasts. Patients with low probability of responding to IST should be treated with azacitidine, decitabine, or lenalidomide. Patients with del (5q) with or without other cytogenetic abnormalities and symptomatic anemia should receive lenalidomide. Studies have shown improved quality of life outcomes in these patients with lenalidomide.22,23 Both azacitidine and decitabine have not been compared with each other or with lenalidomide in any randomized trial in terms of efficacy.
High-risk patients (IPSS intermediate-2, high; IPSS-R intermediate, high, very high; WPSS high, very high) have an estimated median survival of 8–18 months with supportive care alone.14 Hence, intensive chemotherapy or allogeneic (HCT) should be offered to patients with good performance status in an attempt to alter the disease course. Patients who have related or unrelated matched donor, high-intensity chemotherapy followed by allogeneic HCT is recommended. If a donor is not available, these patients may be treated with intensive induction therapy alone such as that used for AML. Patients who are not eligible for transplant, the use of azacitidine, decitabine, or a relevant clinical trial should be considered if available.
There is general lack of effective treatment for the management of recurrent or refractory MDS. Such patients should be encouraged to participate in clinical trials if available.
Role of Hematopoietic Cell Transplantation in Myelodysplastic Syndrome
Allogeneic HCT is the only known cure for MDS patients. When deciding transplantation for MDS patients, factors such as transfusion dependency, cytogenetics, co-morbidities, patient's age, and IPSS-R risk should all be considered.
For those patients with higher risk disease, allogeneic HCT from an HLA-matched sibling donor is the preferred approach. For those without HLA-matched sibling, a fully matched (8 of 8) unrelated donor will result in comparable survival rates, but higher treatment-related mortality. Due to certain factors such as donor availability and advanced age of most patients, the use of allogeneic HCT in MDS is limited. However, the advent of reduced intensity HCT and the use of matched unrelated donors have extended the applicability of HCT to patients in their early 60s and even early 70s.
There have been no prospective trials evaluating the timing of transplantation in MDS patients. However, several observational studies suggest that the patients with higher risk disease are most likely to benefit from allogeneic HCT while patients with lower risk disease are better served by delaying transplantation until progression to higher risk disease, but before transformation to AML.24 Since peripheral blood progenitor cells engraft more rapidly than bone marrow, they are the preferred source of hematopoietic stem cells in MDS patients.
There is little consensus on the appropriate conditioning regimen for the transplant patients. The challenge is that of balancing regimen-related toxicity against relapse risk. The two most common regimens employed are busulfan/cyclophosphamide or cyclophosphamide/total-body irradiation (TBI). Studies have described equal outcomes following conditioning with either of the regimens.
Nonmyeloablative or reduced intensity regimens are potentially an option in patients with advanced age or with co-morbidities. Studies have suggested that such regimens are sufficient enough to induce durable engraftment, tolerance, and graft-versus-tumor effect in the mixed chimeras with reduced graft-versus-host-disease. Such regimens commonly include fludarabine, low-dose TBI, alemtuzumab, or ATG in combination with other agents.
Autologous transplant has been studied in patients for whom allogeneic donors were not available.25,26 However, due to higher incidence of relapse, autologous HCT has been evaluated only in limited numbers of patients and is generally not recommended.
Management of the Complications of Myelodysplastic Syndrome
-
Anemia – At least 80% of patients are anemic at the time of diagnosis.27 Patients should be evaluated for the other causes of anemia. The threshold for red cell transfusion varies with patient age, symptoms, and the presence of medical co-morbidities. Erythropoietic stimulating agents may decrease the need for red cell transfusions in patients with lower risk disease and with sEpo levels ≤500 mU/mL. However, for patients with chronic RBC transfusion need, serum ferritin levels should be monitored
-
Infection – It is the principal cause of death in MDS patients. Empirically broad spectrum antibiotics should be started for patients who are septic and/or have an ANC <500 cells/µ. According to NCCN guidelines, use of prophylactic antibiotics is not recommended
-
Bleeding – It is primarily managed by platelet transfusions, aminocaproic acid, or other antifibrinolytic agents. Thrombopoietin mimetics have not been approved by the US Food and Drug Administration in MDS at the moment
-
Cutaneous lesions – Sweet syndrome and myeloid sarcoma warrant particular attention since they may be indications for more aggressive therapy
-
Transformation to AML, which is often less responsive to standard treatment than is de novo AML.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- SEER Cancer Statistics Review 1975-2010: Section 30 – Myelodysplastic Syndrome (MDS), Chronic Myeloproliferative Disorders (CMD), and Chronic Myelomonocytic Leukemia (CMML); 2012. Available from: http://www.seer.cancer.gov/archive/csr/1975_2010/results_merged/sect_30_mds.pdf [Last accessed on 2016 Feb 12].
- [Google Scholar]
- Myelodysplastic syndromes In: Greer J P, ed. Wintrobe's Clinical Hematology (12th ed.). Baltimore: Lippincott Williams & Wilkins; 2008.
- [Google Scholar]
- Granulocytic sarcoma in myelodysplastic syndromes: Clinical marker of disease acceleration. Am J Med. 1991;90:274-6.
- [Google Scholar]
- Blood neutrophil function in primary myelodysplastic syndromes. Br J Haematol. 1983;55:217-27.
- [Google Scholar]
- Paraneoplastic autoimmune phenomena in patients with myelodysplastic syndromes: Response to immunosuppressive therapy. Br J Haematol. 1995;91:403-8.
- [Google Scholar]
- Paroxysmal nocturnal hemoglobinuria cells in patients with bone marrow failure syndromes. Ann Intern Med. 1999;131:401-8.
- [Google Scholar]
- HLA-DR15 (DR2) is overrepresented in myelodysplastic syndrome and aplastic anemia and predicts a response to immunosuppression in myelodysplastic syndrome. Blood. 2002;100:1570-4.
- [Google Scholar]
- Factors affecting response and survival in patients with myelodysplasia treated with immunosuppressive therapy. J Clin Oncol. 2008;26:2505-11.
- [Google Scholar]
- STAT3 mutations indicate the presence of subclinical T-cell clones in a subset of aplastic anemia and myelodysplastic syndrome patients. Blood. 2013;122:2453-9.
- [Google Scholar]
- WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues Lyon, France: IARC Press; 2008. editors.
- [Google Scholar]
- International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. 1997;89:2079-88.
- [Google Scholar]
- Proposal for a new risk model in myelodysplastic syndrome that accounts for events not considered in the original international prognostic scoring system. Cancer. 2008;113:1351-61.
- [Google Scholar]
- Prognostic relevance of morphological classification models for myelodysplastic syndromes in an era of the revised international prognostic scoring system. Eur J Cancer. 2016;56:10-20.
- [Google Scholar]
- Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120:2454-65.
- [Google Scholar]
- Myelodysplastic syndromes: 2015 Update on diagnosis, risk-stratification and management. Am J Hematol. 2015;90:831-41.
- [Google Scholar]
- The evolving field of prognostication and risk stratification in MDS: Recent developments and future directions. Blood Rev. 2016;30:1-10.
- [Google Scholar]
- Prognostic significance of combined MN1, ERG, BAALC, and EVI1 (MEBE) expression in patients with myelodysplastic syndromes. Ann Hematol. 2012;91:1221-33.
- [Google Scholar]
- Myelodysplastic syndromes: Contemporary review and how we treat. Am J Hematol. 2016;91:76-89.
- [Google Scholar]
- Risk stratification based on both disease status and extra-hematologic comorbidities in patients with myelodysplastic syndrome. Haematologica. 2011;96:441-9.
- [Google Scholar]
- Hypoalbuminemia is an independent prognostic factor for overall survival in myelodysplastic syndromes. Am J Hematol. 2012;87:1006-9.
- [Google Scholar]
- Prognostic models in myelodysplastic syndromes. Hematology Am Soc Hematol Educ Program. 2013;2013:504-10.
- [Google Scholar]
- Health-related quality of life outcomes of lenalidomide in transfusion-dependent patients with Low- or intermediate-1-risk myelodysplastic syndromes with a chromosome 5q deletion: Results from a randomized clinical trial. Leuk Res. 2013;37:259-65.
- [Google Scholar]
- Lenalidomide in international prognostic scoring system low and intermediate-1 risk myelodysplastic syndromes with del (5q): An Italian phase II trial of health-related quality of life, safety and efficacy. Leuk Lymphoma. 2013;54:2458-65.
- [Google Scholar]
- A decision analysis of allogeneic bone marrow transplantation for the myelodysplastic syndromes: Delayed transplantation for low-risk myelodysplasia is associated with improved outcome. Blood. 2004;104:579-85.
- [Google Scholar]
- Feasibility of peripheral blood progenitor cell harvest and transplantation in patients with poor-risk myelodysplastic syndromes. Br J Haematol. 1996;92:351-9.
- [Google Scholar]
- Successful peripheral blood stem cell transplantation for myelodysplastic syndrome. Bone Marrow Transplant. 1999;24:1343-5.
- [Google Scholar]
- The myelodysplastic syndromes: Diagnosis and treatment. Mayo Clin Proc. 2006;81:104-30.
- [Google Scholar]




