

Ewing's Sarcoma of the Gastrointestinal Tract—A Case Series
Address for correspondence Mintu Mathew Abraham, MD, DNB, DM, Department of Medical Oncology, Regional Cancer Centre, Trivandrum, Kerala 695011, India. drmintumathew@gmail.com
This article was originally published by Thieme Medical and Scientific Publishers Pvt. Ltd. and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Ewing's sarcoma is a neoplasm of the undifferentiated small round cells, which generally affects the bone and deep soft tissues of children and adolescents. Extraskeletal Ewing's sarcomas are less common than skeletal Ewing's sarcoma with the most common sites being chest wall, retroperitoneal space, lower extremities, paravertebral region, and gluteal region. Primary Ewing's sarcoma of the gastrointestinal tract is rare. The rarity of cases, lack of specific literature, or guidelines for treatment and varying presentations make these cases challenging to diagnose and to treat. In this series, we share our experience from a tertiary cancer care center in South India. By our knowledge, this is the first case series in literature sharing the experience from a single center in treating four cases of gastrointestinal Ewing's sarcoma.
Keywords
Ewing's sarcoma
extraskeletal
gastrointestinal

Introduction
Ewing's sarcoma (ES) is a neoplasm of the undifferentiated small round cells, which generally affects the bone and deep soft tissues of children and adolescents. Extraskeletal ES has the same genetic origin as ES, typically a 11;22 (q24;q12) translocation, ES gene/friend leukemia integration 1 transcription factor gene (EWS/FLI-1) fusion. Any part of the human body could be affected by these tumors.
Primary ES of the gastrointestinal tract (GIT) is rare. A comprehensive review of literature published in 2018 by Bong et al1 included a total of 58 cases of GI ES/PNET (primitive neuroectodermal tumors) published in English literature till then. Here, we present a series of four cases of GI ES diagnosed and treated at a tertiary cancer care center in India.
Case 1
A 19-year-old boy was evaluated for symptoms of abdominal pain and vomiting of 3 months duration. He had no other symptoms and on examination he had an ill-defined mass lesion involving the left side of abdomen. Contrast-enhanced computed tomography (CT) scan of the abdomen showed a 11 × 8.5 × 9 cm inhomogeneous solid mass lesion anterior to the left kidney arising from the duodenum. He underwent wide excision of the lesion with excision of segment of bowel and lymphadenectomy. Histopathology report (HPR) showed a 5.5 × 4 × 2.5 cm lesion arising from the duodenum with the resected bowel margin involved by the neoplasm. HPR showed fragments of intestine with a neoplasm in the submucosa showing atypical small round cells with bubbly cytoplasm in sheets and lobules. The nucleus was vesicular with fine chromatin with large areas of necrosis. On immunohistochemistry (IHC), the tumor cells were positive for MIC2/CD99 (Cluster of Differentiation 99), vimentin, cytokeratin (AE1/AE3), CD117 and synaptophysin and negative for chromogranin, Leukocyte Common Antigen (LCA), CD20, TLE-1 (Transducin-Like Enhancer of split-1), DOG-1, and CD34. Thus, a diagnosis of ES/PNET of duodenum with epithelial differentiation and CD117 expression was made. Tumor cells were positive for EWSR1 translocation by dual color break apart from fluorescence in situ hybridization (FISH) analysis.
Bone marrow evaluation was within normal limits. Postoperative positron emission tomography-computed tomography (PET-CT) showed a small fluorodeoxyglucose avid soft tissue nodule in the left para-aortic region suspicious of lymph node involvement. His blood counts, liver and renal function tests, and metabolic parameters were within normal limits. His lactate dehydrogenase was 183 U/L
He was given adjuvant chemotherapy with vincristine, Adriamycin, cyclophosphamide alternating with ifosfamide and etoposide (VAC/IE) schedule. Radiotherapy (RT) to local site of 50.4Gy/28# was also given at 12th week in view of margin positivity and suspicious lymph node in PET-CT. He had completed 17 cycles of chemotherapy and the end of treatment PET-CT showed no residual disease. He was asymptomatic on 6 months follow-up.
Case 2
A 17-year-old girl was evaluated with complaints of melena and persistent anemia. Contrast-enhanced CT of the abdomen showed a 2.8 × 2.5 cm lesion arising from the D2 section of duodenum with few smaller nodules in mesentery.
She underwent Whipple's resection. HPR showed a 3.5 × 3.5 × 3 cm lesion infiltrating up to the duodenum with negative margins. Microscopy showed a cellular neoplasm with the neoplastic cells in nests and sheets with round-to-oval shape, scanty cytoplasm, and irregular vesicular nuclei. The tumor cells were positive for cytokeratin, vimentin, CD99 and negative for CD10, synaptophysin, chromogranin desmin, CK7, CK20, calretinin, and CD56. The picture was compatible with ES/PNET of duodenum. Postoperative CT showed no evidence of any residual disease. Bone marrow evaluation was within normal limits. Metastatic workup including bone scan and CT Thorax did not show evidence of any metastasis. Her blood counts, liver and renal function tests, and metabolic parameters were within normal limits.
She was given adjuvant chemotherapy with VAC/IE X 17 cycles. End of treatment evaluation did not show any significant abnormality. She is currently on follow-up and asymptomatic for the last 54 months.
Case 3
A 35-year-old gentleman presented with complaints of constipation and urinary retention for the last3 months duration. Colonoscopy revealed a large mass occluding the lumen of rectum involving the distal and mid rectum with no mucosal ulceration. A magnetic resonance imaging (MRI) of pelvis was done that revealed a 11 × 10 × 8.8 cm mass lesion arising from the wall of the rectum (Fig. 1) and infiltrating the psoas muscle and levator ani, distally reaching up to 4 cm from anal verge.
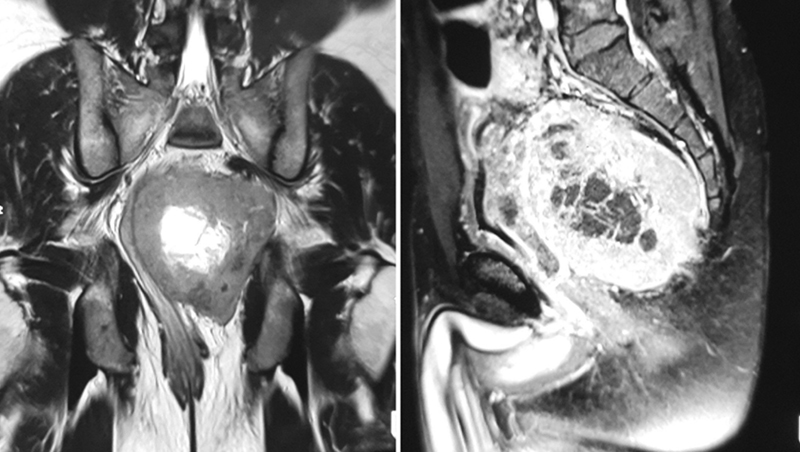
-
Fig. 1 Magnetic resonance imaging scan of the pelvis with T2-weighted coronal and T1-weighted sagittal image of the pelvis showing a large exophytic well-marginated mass arising from the left lateral of the rectum, which is heterogeneous with central necrosis.
Biopsy from the lesion showed a neoplasm composed of small round cells with scanty-to-moderate cytoplasm and round-to-oval nuclei in loosely cohesive nests (Fig. 2A and B). On IHC, the tumor cells were positive for MIC-2 (Fig. 2C), cytokeratin (AE1/AE3), and negative for desmin, myogenin, synaptophysin, chromogranin, and TLE-1. FISH for EWSR1 translocation was positive (Fig. 2D) (by dual color break apart FISH analysis). Bone scan, CT thorax, and bone marrow evaluation were within normal limits. Thus, a diagnosis of ES of rectum was made. Metastatic workup including bone scan, CT thorax, and bone marrow evaluation were within normal limits.
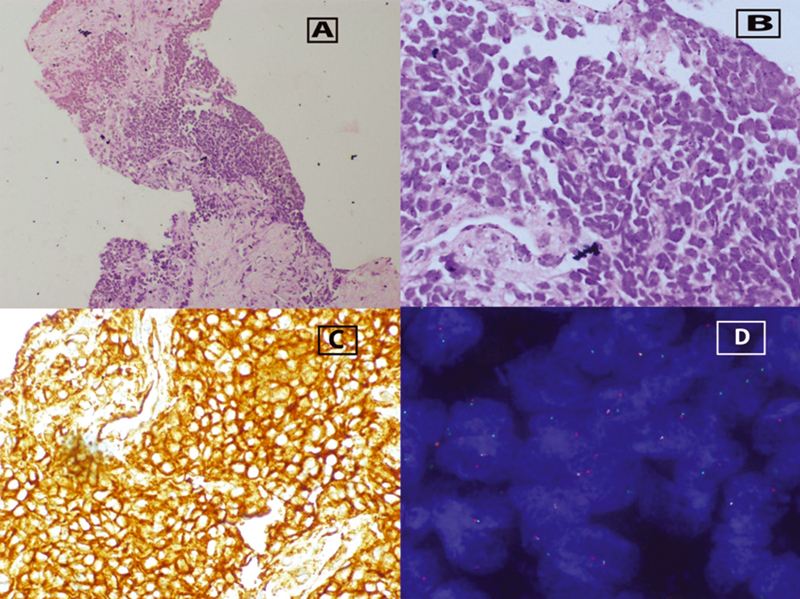
-
Fig. 2 (A) Trucut biopsy rectal growth with nests of small round blue cells. (100x), (B) small round cells showing moderate cytoplasm and round-to-oval hyperchromatic nuclei (400x), (C) immunohistochemistry positivity for MIC-2, and (D) fluorescence in situ hybridization showing break-apart signals indicating EWSR1 gene rearrangement.
He was started on chemotherapy with VAC/IE. After four cycles of chemotherapy, he had rapid worsening of liver function test and altered coagulation profile and was detected to have hepatitis B reactivation. His liver function improved on supportive care and antivirals. Definitive surgery was not feasible and hence he has undergone definitive local RT and is continuing adjuvant chemotherapy at present.
Case 4
A 41-year-old female was evaluated for the complaints of bleeding per vaginum and underwent laparotomy for total hysterectomy in November 2015. Preoperatively, an exophytic growth was found arising from the sigmoid colon and it was also resected. Microscopy of the tumor revealed a malignant small round cell tumor infiltrating the omentum, serosa, and muscularis propria of sigmoid colon. IHC was positive for CD99 and was negative for desmin, myogenin, and LCA. Thus, a diagnosis of ES/PNET of sigmoid colon was made.
Her hematology, liver and renal function tests were within normal limits. Bone scan, CT thorax, and bone scan showed no evidence of disease involvement elsewhere. She was given adjuvant treatment with VAC/IE for 17 cycles. Post chemotherapy re-evaluation was within normal limits. She was on regular follow-up and was asymptomatic for the last 48 months.
Discussion
Extraskeletal ES are less common than skeletal ES with the most common sites being chest wall, retroperitoneal space, lower extremities, paravertebral region, and gluteal region.2 Primary GI origin of ES is very rare. This series of cases shows a single institutional experience in treating this rare site of ES over the past 7 years. The rarity of cases, lack of specific literature or guidelines for treatment, and varying presentations make these cases challenging to diagnose and to treat.
Among four, two patients were males and two were females with an age ranging from 17 to 41 years (Table 1). The presentation was varying depending on the site of the tumor. It may be difficult to determine the exact site of origin of upper GI ES and to differentiate between a pancreatic origin and a duodenal origin due to the tumor infiltrating into nearby organs. In two of our patients, the origin was more likely from the duodenum taking into account the radiological and pathological findings in these patients. In the review by Bong et al,1 pancreas was the site in the majority of cases. Lower GI ES are also very rare. The review of literature of rectal ES by Aboumarzouk et al3 reports a total of only three cases of rectal ES in literature. The review by Parcesepe et al4 reports that colonic ES is also rare with only five cases reported in literature till 2019. One of the patients in our series had origin of lesion from the colon and another had origin from rectum.
|
No of patients |
Age/sex |
Site |
Mets/Non-mets |
Treatment |
Status |
|---|---|---|---|---|---|
|
1 |
19/M |
Duodenum |
Non-mets |
Excision—chemo (VAC/IE x 4)—RT (50.4Gy/28#) - Chemo (VAC/IE for a total of 17 cycles) |
Alive NED at 6 months |
|
2 |
17/F |
Duodenum |
Non-mets |
Whipple's resection—chemo (VAC/IE x 17 cycles) |
Alive NED at 48 months |
|
3 |
35/M |
Rectum |
Non-mets |
Chemo (VAC/IE x 4)—RT (54Gy/30#)—now on chemo |
On chemo |
|
4 |
41/F |
Colon |
Non-mets |
Resection—chemo (VAC/IE x 17 cycles) |
Alive NED at 54 months |
Abbreviations: Chemo, chemotherapy; F, female; M, male; Mets, metastatic; NED, no evidence of disease; RT, radiotherapy; VAC/IE, vincristine, Adriamycin, cyclophosphamide alternating with ifosfamide and etoposide.
Three of the patients underwent wide excision and the HPR revealed the malignancy, while one underwent neoadjuvant chemotherapy after biopsy. The pathological appearance is of small round blue cells with scanty cytoplasm and round-to-oval nuclei. The cells may be positive for CD99(MIC-2) vimentin, cytokeratin (AE1/AE3), and neuronal markers,5 while other IHC markers may be used to rule out clinical and radiological differential diagnosis. Diagnosis is confirmed by the presence of typical mutations detected by FISH or PCR analysis. In 85% of cases, a reciprocal translocation t(11;22)(q24;q12), resulting in EWSR1-FLI1 fusion, can be detected, whereas the t(21; 22)(q22; q12), resulting in EWSR1-ERG fusion, can be found in around 10% of cases. Other translocations can also occur, involving other ETS genes (FEV, ETV1, E1AF).6
The staging evaluation and metastatic workup is derived from that of skeletal ES with local imaging depending on the site of origin of the tumor. In this series, three of the patients underwent contrast-enhanced CT to assess local disease, while the patient with the rectal tumor underwent evaluation with an MRI of the pelvis. PET-CT is a useful tool in staging and detection of residual disease.7 It can also be useful in determining postoperative residual disease and was used in one of our patients to assess the same. Standard metastatic workup for ES including CT thorax, bone scintigraphy, and bone marrow evaluation was also done in all patients.
All these cases were discussed in the institutional multidisciplinary tumor board prior to starting treatment. The importance of a center experienced in treating sarcomas for rare tumors like ES especially for such extremely rare sites has been reflected in their superior outcome after treatment in such centres.8 Three of the patients in this series underwent unplanned resections outside hospitals. One of the operated patients had to receive radiation post four cycles of chemotherapy as it was an incomplete surgical excision.
There are no standard guidelines for the treatment of extraosseous ES and protocols used for skeletal ES are generally followed. All the patients in this series received standard chemotherapy for ES with VAC/IE schedule for 17 cycles and tolerated chemotherapy well. Two patients are on long follow-up without any significant toxicities.
The review from St. Jude hospital by Orr et al9 shows that the survival in extraosseous ES is equivalent to that of skeletal ES. There is paucity of literature to comment on the survival in GIT ES. In our series, two patients are on follow-up for more than 4 years; one patient recently completed treatment and one patient is on active treatment now.
Conclusion
GI system is a rare site for ES. The presentation may vary but radiology, histopathology, IHC, and molecular studies contribute to diagnosis. Guidelines for skeletal ES modified according to the site of disease may be used for evaluation and treatment. Two patients in the series have long-term survival without significant toxicities. A multidisciplinary treatment approach at a high-volume center should be considered for rare diseases like GIT ES.
Conflict of Interest
None declared.
References
- Case report and literature review of Ewing's sarcoma in the gastrointestinal tract. Surg Pract. 2018;22(02):84-92.
- [Google Scholar]
- Primary extraskeletal Ewing's sarcoma/primitive neuroectodermal tumor of breast. Indian J Radiol Imaging. 2016;26(02):226-230.
- [Google Scholar]
- PNET/Ewing's sarcoma of the rectum: a case report and review of the literature.
- Colonic Ewing Sarcoma/PNET associated with liver metastases: a systematic review and case report. Pathol Res Pract. 2019;215(02):387-391.
- [Google Scholar]
- Pathology of Ewing's sarcoma/PNET: current opinion and emerging concepts. Indian J Orthop. 2010;44(04):363-368.
- [Google Scholar]
- Bone sarcomas: ESMO-EURACAN-GENTURIS-ERN PaedCan Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann Oncol. 2021;32(12):1520-1536.
- [Google Scholar]
- The importance of PET/CT in the evaluation of patients with Ewing tumors. Radiol Bras. 2015;48(03):175-180.
- [Google Scholar]
- Ewing sarcoma-diagnosis, treatment, clinical challenges and future perspectives. J Clin Med. 2021;10(08):1685.
- [CrossRef] [Google Scholar]
- Analysis of prognostic factors in extraosseous Ewing sarcoma family of tumors: review of St. Jude Children's Research Hospital experience. Ann Surg Oncol. 2012;19(12):3816-3822.
- [Google Scholar]




