

Translate this page into:
Ewing’s Sarcoma of the Cranial Vault Revealed by Macrocrania
Address for correspondence Abdoulaye Diop, Assane Seck University, Ziguinchor, Dakar 523, Senegal. a.diop@univ-zig.sn
This article was originally published by Thieme Medical and Scientific Publishers Private Ltd. and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Ewing’s sarcomas are a heterogeneous group of aggressive tumors affecting mostly children and young adults. They account for 10% of primary malignancies of the bones and 3% of all malignancies in children. They mainly affect the long bones, pelvis, and ribs. Cranial vault localization is extremely rare and indeed account for less than 1% of all localizations in the body. We are reporting the unusual case of a 3-year-old child who presented to our unit with a macrocrania which had been evolving for 2 months prior to his admission, associated with sporadic vomiting and a right frontal swelling that was gradually installed. The cerebral computed tomography scan showed a right frontoparietal lytic tissular process, heterogeneously enhanced after injection of contrast material; with dural invasion and intraparenchymal extension. The child was biopsied and the pathological examination concluded Ewing’s sarcoma. The evolution was fatal with the passing of the child 2 weeks after the biopsy.
Keywords
Ewing’s sarcoma
cranial vault
macrocrania

Introduction
Ewing’s sarcoma is the second most prevalent bone tumor in children after osteosarcoma.1 It belongs to a large family of small blue round cell tumors. Ninety percent of these sarcomas occur in the first or second decade of life.2 The most affected sites are long bones, ribs, and the pelvic belt.3,4 The cranial vault is rarely affected, representing 1% of all sites.1,5 The symptomatology is usually atypical and nonspecific. We are reporting an unusual case of macrocrania revealing a Ewing’s sarcoma of the cranial vault, with dural invasion and intracerebral extension in a 3-year-old child.
Case Report
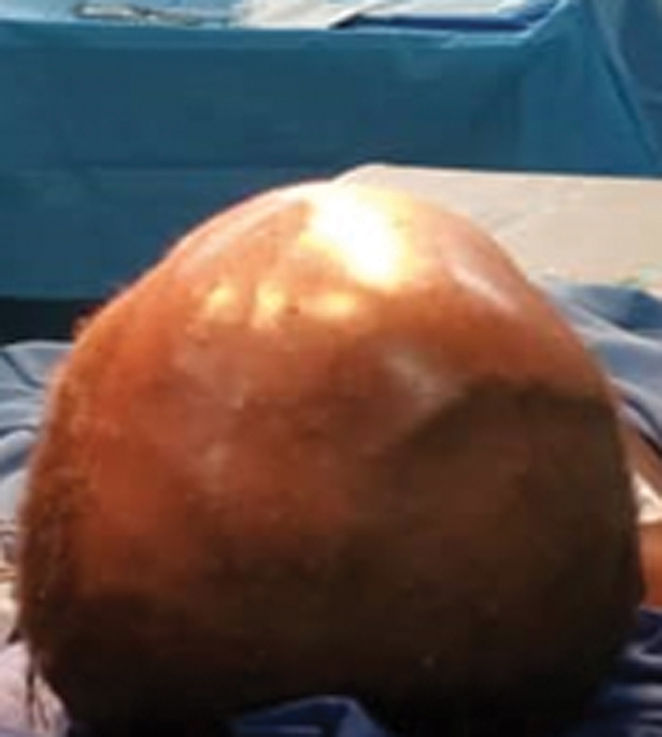
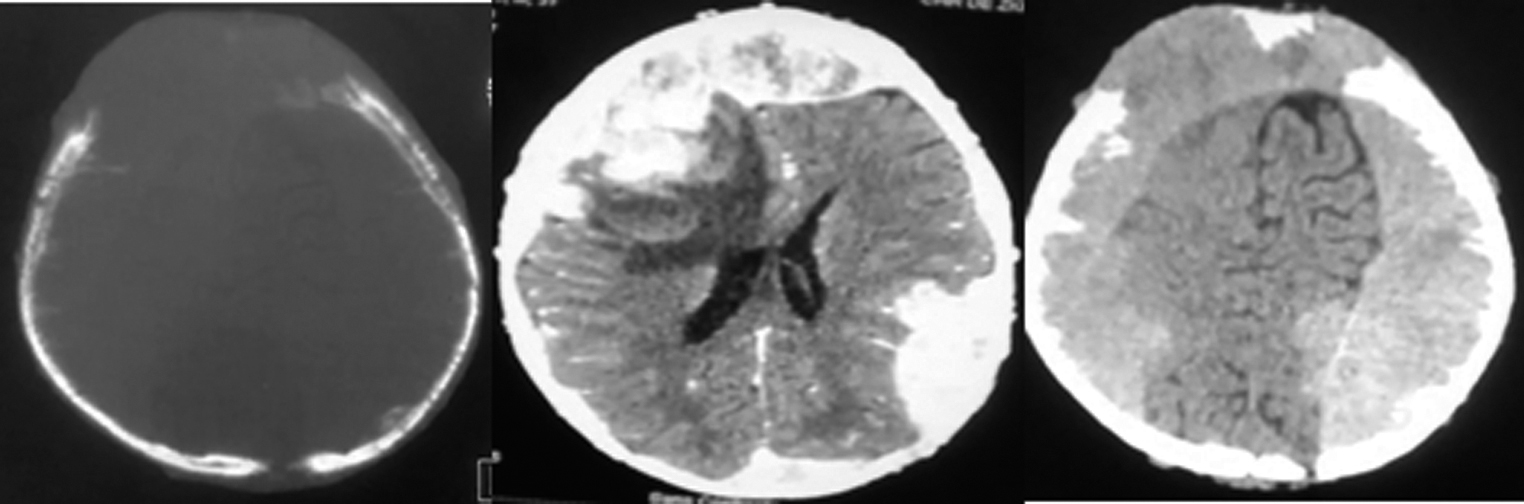
The patient was a 3-year-old boy with no notable pathological history. He presented to our unit with a macrocrania (Fig. 1) which had been evolving for 2 months prior to his admission and sporadic vomiting. Neurological examination showed altered consciousness with a Glasgow Coma Scale rated to 12 (E3M5V4), an intracranial hypertension syndrome, and a proportional left hemiparesis. The examination of the cephalic extremity showed a macrocrania with a cranial perimeter of 65 cm and a right frontal swelling, fixed to the vault of the skull, long axis measuring 40 mm, with a healthy skin above. The child was also clinically anemic and severely malnourished. The rest of the clinical examination was within normal limits. The brain computed tomography (CT) scan (Fig. 2) showed a right frontoparietal lytic tissular process which was heterogeneously enhanced after injection of contrast material. It has a bilateral hemispherical endocranial extension mainly in the extradural area, as well as dural invasion, intraparenchymal extension with significant perilesional edema. This exerts a mass effect on the lateral ventricles which made them collapse, and also on the midline which deviated to the left. The process seemed to originate from the vault which was lysed, along with infiltration of the soft tissues.
-
Fig. 1 Macrocrania with right frontal swelling (preoperative).
-
Fig. 2 Cerebral computed tomography scan showing a lytic, right frontoparietal tissular process heterogeneously enhanced after injection of contrast material with bilateral hemispherical predominantly extradural endocranial extension with dural invasion and intraparenchymal extension and significant perilesional edema.
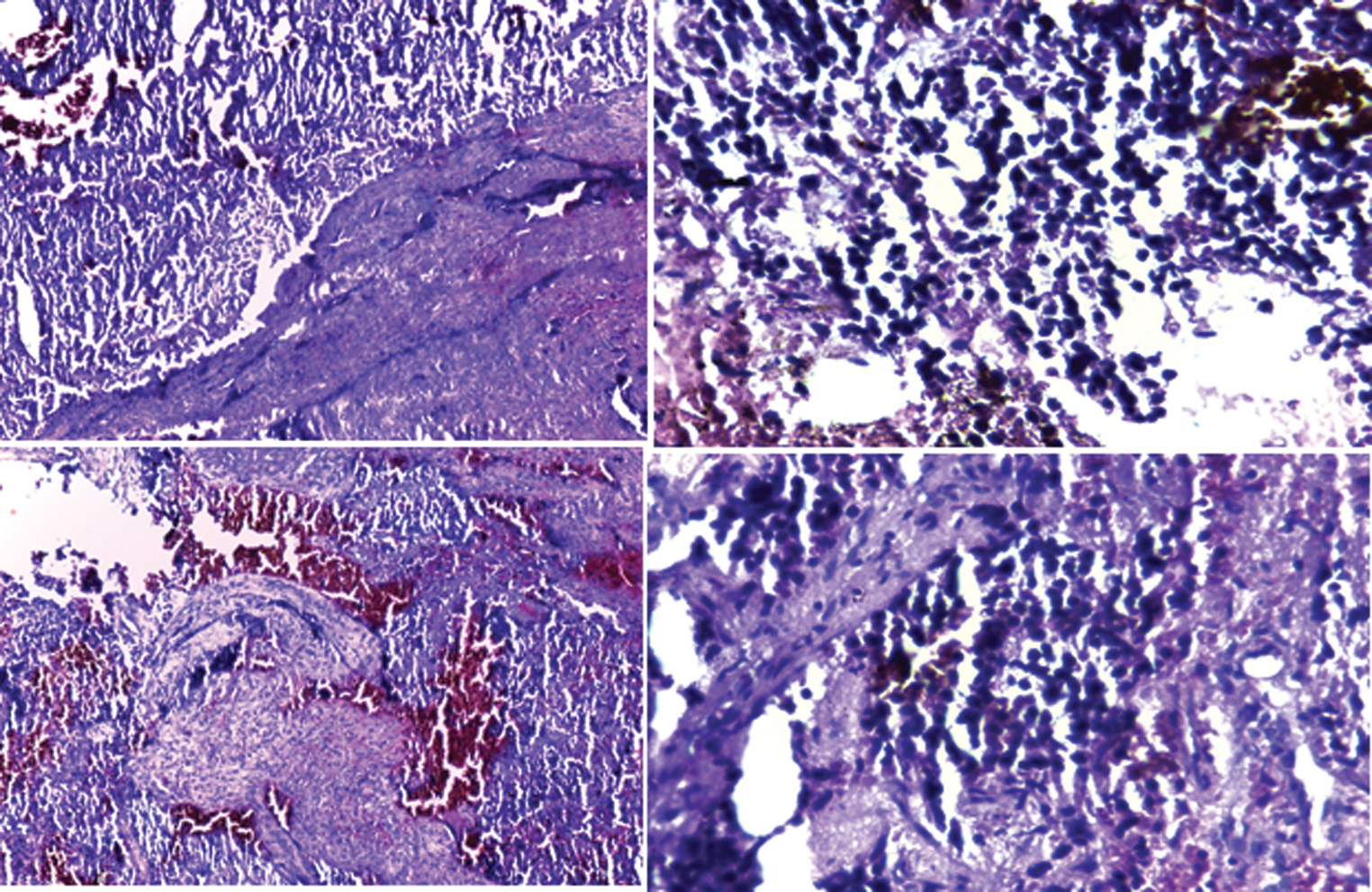
Chest, pelvis, and both femurs X-rays and abdominal ultrasound were performed as a part of workup for extension and returned normal. The child was biopsied using a paramedian frontal approach focused on the swelling. While operating, we found out a grayish fleshy and very hemorrhagic tumor with a completely eroded frontal bone. We collected samples of the eroded bone as well as the invaded dura mater. Anatomopathological examination confirmed Ewing’s sarcoma with a proliferation of small round cells of compact layered architecture with slight neovascularization of the sinusoid type (Fig. 3). The evolution was fatal, with the passing of the child during hospitalization, 2 weeks after the biopsy.
-
Fig. 3 Proliferation of small round cells into compact sheet architecture with slight sinusoid-like neovascularization.
Discussion
Described in 1921 by James Ewing, Ewing’s sarcoma is the most common primary malignant bone tumor after osteosarcoma in young people.1 In 90% of cases it occurs in the first or second decade of life with a slight male predominance.1,2 These tumors mostly involve the long bones, pelvis, or ribs.3 Primary cranial localization is rare and accounts for only 1% of sites.1,5 Pritchard et al found only 3 cranial cases out of 229.6 However, the frontoparietal location described in our study is the most frequently found in the literature.5
The symptomatology is usually atypical and nonspecific, as symptoms appear depending on dural invasion, hydrocephalus, or increased intracranial pressure. It is the localized swelling of the arch that catches attention.7 It rapidly evolves, and is attached to the bone, as in our case. The signs of intracranial hypertension and motor deficit found in our patient seemed to be related to the volume of the tumor and its intracranial extension. No cases of Ewing’s sarcoma with endocranial extension revealed by macrocrania have ever been reported in the literature. The pattern of a secondary and progressive macrocrania in children related to a tumor process remains that of the choroid plexus papilloma. However, any supra- or subtentorial tumor can induce intracranial hypertension with progressive macrocranial involvement.8
The bone window cerebral CT scan remains the gold standard examination, and allows a better analysis of bone lesions as well as intra- and extracranial extension.1,2 Our patient had a large tumor with bilateral hemispherical endocranial extension mainly in the extradural area, which may be confused with an epidural hematoma. This multifocal extradural intracranial location has been described by Wang and Guo.9
Ewing’s sarcoma is a highly aggressive and rapidly metastatic tumor.10 However, early metastases are less common in primary Ewing’s sarcoma, so cranial localizations are considered to have a better prognosis.11,12 Unlike what has been reported in the literature, the evolution of our patient was fatal due to the size of the tumor and its intracranial invasion. The extent of the endocranial invasion and the soft tissue of the tumor made it impossible to perform total or partial excisional surgery in that case. Chemotherapy and radiotherapy are the treatment of choice in inoperable cases. Our patient passed away long before the histological confirmation of the diagnosis, and thus, could not benefit from further treatment.
Conclusion
Ewing’s sarcoma is a malignant bone tumor of the young subject. Primary localization at the vault of the skull is rare. The signs are mostly atypical and nonspecific. However, macrocrania as a circumstance of discovery of the primitive localization of the sarcoma of the cranial vault is exceptional.
Conflict of Interest
None declared.
References
- Primary Ewing sarcoma of the skull vault [in French] J Radiol. 2007;88(12):1899-1901.
- [Google Scholar]
- Primary Ewing sarcoma on skull vault in a child. Ind J Radiol Imag. 2003;13:303-305.
- [Google Scholar]
- Ewing’s sarcoma of the hand following recurrent trauma; a case report. Hand. 1980;12(03):300-303.
- [Google Scholar]
- Ewing’s sarcoma. A clinicopathological and statistical analysis of patients surviving five years or longer. J Bone Joint Surg Am. 1975;57(01):10-16.
- [Google Scholar]
- Primary Ewing’s sarcoma of the occipital bone presenting as hydrocephalus and blindness. Pediatr Neurosurg. 2007;43(02):170-173.
- [Google Scholar]
- Children with macrocrania: clinical and imaging predictors of disorders requiring surgery. AJNR Am J Neuroradiol. 2001;22(03):564-570.
- [Google Scholar]
- Multiple primary Ewing’s sarcomas in cerebral cranium of a child: a case report and review of the literature. Int J Clin Exp Pathol. 2015;8(06):7575-7582.
- [Google Scholar]
- Imaging characteristics of primary cranial Ewing sarcoma. Pediatr Radiol. 2005;35(06):612-618.
- [Google Scholar]
- Primary Ewing’s sarcoma of the cranium. Neurosurgery. 2000;46(01):62-68. , discussion 68–69
- [Google Scholar]
- Ewing’s sarcoma of the skull in an infant. A case report and review. Pediatr Neurosurg. 1996;25(02):100-104.
- [Google Scholar]




