

Translate this page into:
Efficacy of PARP Inhibitors in the Treatment of Ovarian Cancer: A Literature-Based Review
Address for correspondence Venkata Pradeep Babu, MD, DNBRoom 2258, 2nd Floor, C Block, Rajiv Gandhi Cancer Institute and Research Centre, Sector 5, Rohini, New Delhi 110085, India. pradeepbabu.koyyala@gmail.com
This article was originally published by Thieme Medical and Scientific Publishers Private Ltd. and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Poly (ADP-ribose) polymerase (PARP) inhibitors are a unique class of therapeutic agents that focus on tumors with deficiencies in the homologous recombination DNA repair mechanism. Genomic instability outlines high-grade serous ovarian cancer, with 50% of all tumors displaying defects in the important DNA repair mechanism of homologous recombination. Earlier research studies have demonstrated considerable efficiency for PARP inhibitors in patients with germ line breast-related cancer antigens 1 and 2 (BRCA-1/BRCA-2) mutations. It has also been observed that BRCA wild-type patients with other defects in the homologous recombination repair mechanism get benefited from this therapy. Companion homologous recombination deficiency (HRD) scores are being developed to guide the selection of patients that are most likely to benefit from PARP inhibition. The selection of PARP inhibitor is mainly dependent upon the number of prior therapies and the presence of a BRCA mutation or HRD. The identification of cases which are most likely to get benefited from PARP inhibitor therapy in view of HRD and other biomarker assessments is still challenging. The purpose of this review is to focus and describe the current evidences for PARP inhibitors in ovarian malignancy, their mechanism of action, and the outstanding issues, including the rate of long-term toxicities and the evolving resistance.
Keywords
BRCA
PARP inhibitors
resistance mechanism
companion diagnostic
homologous recombination

Introduction
Inhibitors of poly (ADP-ribose) polymerase (PARP) are a type of cancer drugs that are mainly used in the therapeutic management of ovarian cancer (OC). They are also under trials as a treatment for other types of cancer. PARP is a protein detected in cells. It helps the damaged cells in repairing themselves. PARP inhibitors (PARPis) stop the PARP from doing its repair work in cancer cells, which further results in the death of cancer cells. Researchers first focused on these drugs in cancers that already had problems in repairing cell damage. They focused on cancers with a change (or fault) in genes called breast-related cancer antigens (BRCA). Normally, BRCA1 and BRCA2 genes play a vital role in cell repair. Cells are less likely to repair themselves if there is a defect in one or both of these genes. People who have defective BRCA genes have an increased risk of certain cancers including OC, breast cancer, and prostate cancer.
Cancer cells with BRCA gene faults already have a poor repair mechanism. So blocking PARP with a PARPi drug means that the cells are not able to repair themselves and ultimately die. Inhibition of PARP proteins, which a cell utilizes in different types of DNA repair mechanisms, is a promising therapy for OC.1 As OC cells always possess pre-existing defects in damaged DNA repair pathways,2 the inhibition of PARP can result in preferential death of cancer cells when another mechanism for repairing DNA is defective. This is called synthetic lethality. The concept of synthetic lethality is that cell death occurs when two separate mechanisms for repair of defective DNA are present in a cell and both are incapacitated. For example, synthetic lethality occurs when OC cells with a BRCA mutation are exposed to a PARPi. The mutation causes defective repair of breaks in double-stranded DNA and the drug prevents repair of breaks in single-stranded DNA. Thus, the cancer cells that have BRCA mutations die while cells with a functioning BRCA gene remain unaffected.1,2
Two PARPis, olaparib and rucaparib, received United States Food and Drug Administration (FDA) recognition in 2014 and 2016, respectively, for the management of BRCA-associated OC.3 Since 2017, three PARPis—niraparib, olaparib, and rucaparib—have received FDA approval in the recurrent setting as maintenance therapy following platinum-based therapy.4 The approval of these three PARPis in rapid succession has resulted in a paradigm shift in the management of recurrent OC. Additional PARPis include veliparib (ABT-888), talazoparib (BMN-673), pamiparib (BGB-290), iniparib (BSI-201, SAR240550), INO-1001, ABT-767, CEP-9722, E7016/ GPI-21016, and 2X-121. However, these treatment choices present a challenge for clinicians, who now struggle for the selection of PARPis for individual patients, as well as with how to best incorporate these agents into existing treatment protocol.
Among unselected patients with breast cancer, ∼10% harbor a germ line mutation in BRCA1 or BRCA2 genes.5 These are tumor suppressor genes mainly responsible in the maintenance of genome integrity with one of the major DNA damage repair pathways called homologous recombination.6 In the presence of PARPis, the PARP-dependent DNA repair system cannot be activated with consequent development of double-strand breaks. In normal cells, these breakages can be repaired through the homologous recombination pathways with subsequent retrieval of DNA integrity and cell survival, whereas in BRCA-mutated cells, homologous recombination is defective and these damages cannot be efficiently repaired further resulting in cell deaths.7 Preclinical studies demonstrated that BRCA1/2-deficient cancer cells are sensitive to PARP inhibition, due to the persistence of DNA lesions that would be commonly repaired by the homologous recombination pathway.8 Depending on this strong biological rationale, the study by Tutt and colleagues then provided the proof of concept for the potential clinical utility of single-agent PARPi in patients with BRCA-mutated metastatic breast cancer.9 Following these results, over the past few years, multiple clinical trials have studied and are currently investigating the role of different PARPi in this population.
Recently, depending on the results of the phase III randomized controlled clinical trials (RCT) OlympiAD,10 single-agent PARPi olaparib has been approved as the first targeted therapy for the patients with BRCA-mutated HER2-negative metastatic breast cancer.10
Epidemiology of OC
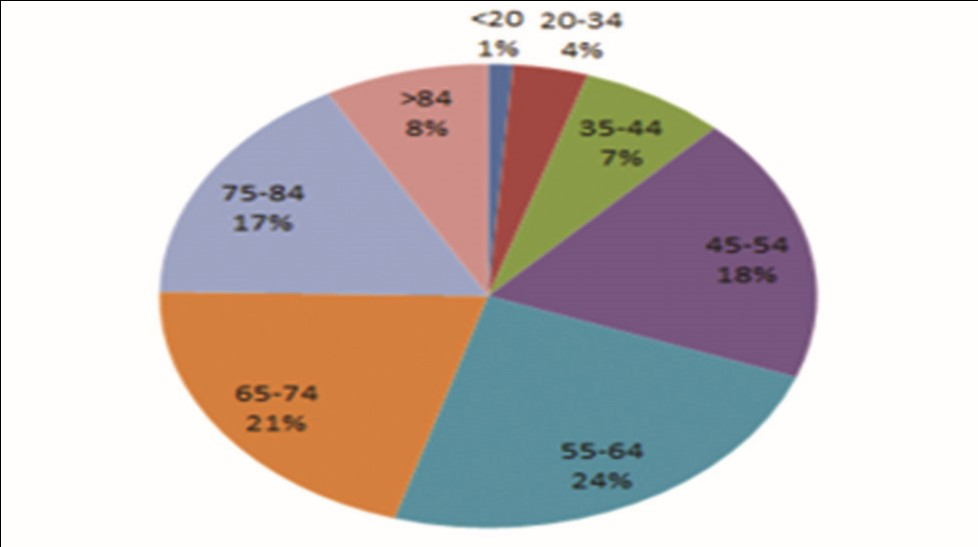
Cancer of ovary is the fifth most leading cause for cancer-related mortality among the women population in the United States.11 The chances of developing OC increases with age (the majority of patients are diagnosed between the age of 55 and 64 years), with the highest mortality rate reported between the age of 65 and 74 years.12 The lifetime risk factors for the development of OC are ∼1 in 77 (1.3%) women (Fig. 1).13 There is a 5-year survival rate of 47% reported among all races with ovarian neoplasm, with a much greater survival rate of 93% reported with localized disease. Comparatively, there is higher rate of mortality and incidence rate of OC in non-Hispanic white women.13 OC cases and mortality are also found to have decreased in the last few years. According to the Center for Disease Control and Prevention, multiple risk factors are found to be responsible for development of OC.12 These factors include family (first-degree relative) history of OC, advanced age, genetic mutations, Eastern European or Ashkenazi Jewish background, personal history of colorectal/uterine/breast cancer, nulliparity, endometriosis, and long-term (more than 10 years) estrogen use without any concomitant use of progesterone. In the United States, around 10 to 15% of OC cases are considered to occur because of germ line BRCA-mutation (gBRCAm)14 (Fig. 2).

-
Fig. 1 Risk factors for ovarian cancer. BRCA, breast-related cancer antigens.
-
Fig. 2 Percentage of ovarian cancer cases diagnosed by age group (in years).
Certain factors have been also found to be related with a diminished risk of developing OC. These factors include certain surgical procedures (e.g., hysterectomy, oophorectomy, and tubal ligation),14 combined use of oral contraceptive for the period of greater than 5 years, childbirth, and breastfeeding for the period of more than 1 year. Primary peritoneal cancers and fallopian tube cancers are always subgrouped with OC, but they are quiet rarely reported. These type of cancers have also been reported to be related with BRCA mutations.15
Genetic Considerations
If personal or family history is suggestive of any possible harmful mutation, patients should consult a health care provider. Screening tools evaluate multiple associated risk factors, which include early diagnosis of breast cancer, family or personal history of breast and OC, bilateral breast cancer, family history of multiple breast cancers, male breast cancer, Ashkenazi Jewish ethnicity, and family history of BRCA1- or BRCA2-related cancers.16 Genetic counseling and testing are advised for adult patients who have comparatively higher chances of developing a BRCA-related malignancy.17
It would be important to have a clear overview on potential harms and benefits with the use of PARPis in terms of expected clinical outcomes, risk of developing adverse events (AEs), and impact on quality of life (QoL). Hence, we planned and conducted a systematic review of literature to better understand and elucidate the role of single-agent PARPi in the therapeutic management of patients with OC.
Objective
The objective was to review the available literature on PARPis and compare the benefits versus risks of PARPis in the treatment of OC.
Importance of this Review
Recently, many new novel biological agents that act in multiple ways to conventional chemotherapy have been discovered. It is, therefore, very much needed to establish whether the addition of these new drugs to conventional chemotherapy is fruitful, in terms of survival and, if so, at what cost, in terms of additional AEs. As these compounds may be reported less toxic as compared with conventional chemotherapeutic agents, it may be feasible to advise these new therapeutic agents in patients who are not currently taking chemotherapy, to reduce the chance of, or delay, the recurrence of their OC.
PARPis and Synthetic Lethality
PARPs regulate a multiple biological mechanisms. PARP-1: Part of the PARP protein family; it is also found to be playing an important role in base excision repair mechanism (BER). DNA modifications induced exogenously or endogenously, can be repaired by BER. After eradication of the damaged base, single strand break (SSB) is produced.18 Usually, PARP-1 binds to the SSB and further attracts other proteins to start with the SSB repair.19 Previously, it was considered that the inhibition of PARP-1 would further lead to the stalling of the replication fork at the SSB,20 further leading to the accumulation of double strand breaks (DSBs) in duplicating cells. Recent findings are also suggesting that some PARPis might also “trap” PARP1 on DNA and thereby further interfere with the catalytic cycle of PARP.21 The capability of PARPis to trap PARP1 on DNA has been found to add to the observed cytotoxicity.22 More recent data demonstrated that PARP also has a functional role in the repair mechanism of DSBs.23 The efficiency of PARPis in BRCA1- and BRCA2-related tumors is dependent on the concept of synthetic lethality, whereas a deficiency in either one of two genes has no prominent effect on the viability of the cell but the combination of defects in both the genes will result in death of the cell.24
Thus, especially in BRCA-deficient and homologous recombination-deficient (HRD) cells, inhibition of PARP enzyme will further lead to the death of the cell. Comparatively, normal cells have a sufficient homology directed repair (HDR) function and therefore they survive PARPi therapy. This further leads to a more precise and less toxic therapy as compared with chemotherapy.22 Therefore, PARPis are considered as a potent medication, especially in BRCA-mutated types of malignancies and other HDR-deficient cancers.25
DNA Repair and BRCA
In past few years, dramatic advances have been reported in understanding about the mechanism and regulation of cellular components that are of vital importance in the repair of damaged DNA. DNA faces multiple types of assaults on its basic native structure and sequence throughout the life of a cell.25 Human cells possess minimum five primary pathways of DNA repair, which are systems that serve to probe and identify defects protecting the genome. The major DNA repair pathways are direct repair pathways, mismatch repair (MMR), base excision repair (BER), nucleotide excision repair (NER), and double-strand break (DSB) recombinational repair, which includes both nonhomologous end-joining (NHEJ) and homologous recombinational repair.26 Reduction, dysfunction, or absence of proteins committed to these pathways may further lead to disastrous cellular consequences, leading to the mutagenesis and toxicity.
Recently, BRCA-1 and BRCA-2 tumor suppressor genes have been corelated with a fundamental role in the response to cellular damage through the activation of specific DNA repair mechanism. Both BRCA1 and BRCA2 proteins are usually found in stable interaction, suggesting co-function of these proteins in pathways of tumor suppression. Both these genes have been proposed to function in DNA repair mechanism and as transcriptional regulators. BRCA-1 and BRCA-2 form a complex with Rad51, a protein with an established role in homologous recombination.27
It has been found that BRCA-1 is also involved in forming a complex with and activation of p53.28 The tumor suppressor protein p53 is involved in different types of human malignancies29; the normal function of p53 is to signal the occurrence of DNA damage and temporarily arrest the cell cycle to either allow repair or trigger cell death. In-depth analysis of the effects of BRCA genes and their transcriptional functions may further result in a clearer understanding of their tissue-specific actions.
BRCA Mutations and Risk of Cancer
Established association is found between germ line mutations in BRCA-1 and BRCA-2 and the development of breast or ovarian cancer syndrome.30 BRCA-1 and BRCA-2 gene mutations are linked to inherited breast and ovarian cancers, and are also reported in sporadic malignancies. These genes can, therefore, be associated with the further development of tumors with mutations derived from either germ line or somatic (tumor only) variants.31
Presently, the method used for the identification of BRCA gene mutations is dependent on DNA sequencing techniques. At present, one of the difficult issues with this method is differentiation between clinically considerable changes and benign nonpathogenic variations in these genes, termed as variants of unknown significance (VUS). Genetic testing has reported that near ∼13% of BRCA-1 and BRCA-2 mutations are VUS, implying clinical uncertainty and ambiguity in risk assessment of tested individuals.32,33 Task of accurately identifying carriers of BRCA mutations is complicated by continued lack of understanding of the significance of different polymorphisms in these genes and the mechanism of tumorigenesis conferred upon mutation.
It has been well recognized that BRCA-1–related breast malignancies are likely to be ER-negative than are BRCA-2 and non–BRCA-1 cancers.34 Estrogen has a profound impact on both normal and malignant cells. Certain genes regulating growth are heavily affected by the effects of estrogen. Breast and ovarian cancers are always initially evaluated for estrogen receptor (ER) status, with the rationale of individualized therapy. Knowledge and status of ER provides additional information in respect to prognosis of patient and treatment directives. Failures of BRCA functioning and estrogen signaling among other mechanisms further promotes a lack of proper DNA surveillance, leading to tumorigenesis. It has been demonstrated that gene silencing of BRCA-1 is found to be associated with increased gene expression of aromatase catalyzing the conversion of steroids into active estrogens. This further results in increased activity and hence increased estrogen production.35 Detailed evaluation of estrogen signaling is mandatory to provide the effective therapeutic, preventive, and possibly curative measures in both BRCA and non-BRCA cancers.
Clinical Development of PARPis
At present, many PARPis are in clinical trial phase either as monotherapy and/or as combination therapy (Table 1). Initially, the European Medical Association (EMA) and the FDA registered olaparib as a maintenance therapy of platinum-sensitive relapsed gBRCAm high-grade serous epithelial ovarian cancer (EOC) that responded very well to platinum-based chemotherapy.36 Recently, olaparib was registered by the EMA irrespective of BRCA status.37 The FDA also registered olaparib as monotherapy for treating patients with gBRCAm recurrent EOC after receiving three or more prior lines of chemotherapy. Additionally, the FDA also registered olaparib for the therapeutic management of patients with gBRCAm, HER2-negative metastatic breast cancer who have previously been managed with chemotherapy. In 2016, rucaparib was registered by the FDA as monotherapy for the management of patients with BRCA-mutated recurrent EOC who have received two or more chemotherapies38 and in 2018 rucaparib was registered as maintenance therapy for patients with recurrent EOC who are in complete or partial response to platinum-based chemotherapy.39 In 2017, niraparib was registered as a maintenance therapy for adult patients with recurrent platinum-sensitive EOC who are in a complete or partial response to platinum-based chemotherapy, irrespective of BRCA status of the tumor.39
|
Drug |
Company |
Administration route |
|---|---|---|
|
O-9201 (olaparib) |
Astra Zeneca |
Oral |
|
PF-0137 (rucaparib) |
Clovis/Pfizer |
IV/Oral |
|
ABT 888 (veliparib) |
Abbott |
Oral |
|
INO-1001 |
Inotek |
IV |
|
GP1201 |
Eisai |
Oral |
|
CEP 9722 |
Cephalon |
Oral |
|
MK 4827 (niraparib) |
Merck/Tesaro |
Oral |
|
BMN 673 |
BioMarin |
Oral |
Abbreviations: IV, intravenous; PARP, Poly (ADP-ribose) polymerase.
Olaparib (AstraZeneca)
Olaparib is one of the most focused and investigated PARPis. It was the first PARPis to investigate pharmacokinetics and pharmacodynamics in patients with, among others, breast and ovarian malignancies. Among the 60 patients enrolled, 22 had a BRCA-1 or BRCA-2 germ line mutation. Results showed a maximum tolerated dose (MTD) of olaparib 400 mg BID. Reported adverse events were mainly of grade 1 and 2 and included vomiting, nausea, altered taste, fatigue, and anorexia. Subsequent phase II clinical trials in patients with a gBRCAm showed an overall response rate (ORR) of 41% in patients with advanced breast malignancies40 and of 33% in recurrent EOC41,42 demonstrated a tumor response rate in metastatic breast cancer patients with ≥ 3 chemotherapy regimens of 12.9% (95% confidence interval [CI], 5.7–23.9). For platinum resistant relapsed EOC, the ORR was 31.1% (95% CI, 24.6–38.1). Depending on these results, a subsequent clinical trial (study 19) was started by Ledermann et al. This phase II clinical trial compared the efficiency of olaparib to placebo as maintenance therapy after response to platinum-based chemotherapy in 265 patients with platinum-sensitive recurrent serous EOC. Overall results demonstrated that olaparib increased median progression-free survival (PFS) as compared with placebo (8.4 vs. 4.8 months, respectively; hazard ratio (HR) 0.35; p < 000.1).43 The subgroup analysis by overall BRCA mutation (BRCAm) status (germ line and somatic) demonstrated a considerable benefit in median PFS in the olaparib group as compared with the placebo group (11.2 vs. 4.3 months, respectively; HR 0.18; p < 0.0001).44 Reported adverse events were mostly of grades 1 and 2. This study finally led to the EMA and FDA registration of olaparib for EOC.9
Veliparib (Abbvie)
Veliparib is another PARPi of which clinical studies reported promising results in recurrent EOC. The first phase I clinical trial by Puhalla et al43 supported for a phase 2 dose (RP2D) of 400 mg BID for the management of platinum-resistant or -refractory EOC or basal-like breast cancer. Sixty out of 88 patients had a gBRCAm. Fatigue, nausea, and lymphopenia were the most commonly reported all-grade toxicities. In phase II clinical trial by Coleman et al,40 veliparib monotherapy was given to 50 patients with persistent or recurrent EOC carrying a BRCAm. Results demonstrated an ORR of 26% with acceptable toxicity.
Niraparib (Tesaro)
Niraparib is another potent PARPi. This PARPi was first time clinically tested in phase I clinical trial by Sandhu et al.45 The RP2D was 300 mg/day with reported adverse events of nausea, anorexia, anemia, fatigue, neutropenia, thrombocytopenia, constipation, and vomiting (grades 1 and 2). Efficiency evaluation showed that 40% of patients with BRCAm recurrent EOC and 50% of patients with metastatic breast cancer had a partial response. Recently, the placebo-controlled phase III clinical trial NOVA,46 with niraparib as maintenance therapy after completing or near complete response to platinum-based chemotherapy in patients with platinum-sensitive recurrent EOC, showed a higher PFS in the niraparib group as compared with placebo group. Patients with a gBRCAm had the largest benefit with an increase in PFS of 21.0 versus 5.5 months (HR: 0.27), followed by a subgroup with HDR deficiency as defined by the My Choice HDR deficiency test (Myriad Genetics, Salt lake City, Utah, United States) with an increase in PFS of 12.9 versus 3.8 months (HR: 0.38). Even in the HDR-proficient group a PFS benefit of 6.9 versus 3.8 months was noted (HR: 0.58). OS data were not mature data. Finally it was concluded that niraparib is beneficial in all patients with platinum-sensitive recurrent EOC in response to platinum-based chemotherapy regardless of the BRCA mutation or HDR deficiency status. Furthermore,47 it was concluded that patients managed with niraparib after complete or partial response can maintain their quality of life.
Rucaparib (Clovis Oncology)
One of the first phase I clinical trial focusing rucaparib was conducted by Kristeleit et al.48 Results demonstrated a RP2D of 600 mg BID10 subsequently, efficacy of intravenous (IV) intermittent and oral continuous dosing schedules of rucaparib in gBRCAm recurrent ovarian and metastatic breast cancer was investigated. The IV intermittent dosing schedule resulted in an ORR of 2%, which was 15% for oral rucaparib. Forty-one percent of the patients on the IV intermittent dosing schedule achieved stable disease (SD) for ≥12 weeks. In the oral continuous dosing cohort 81% patients achieved RECIST (Response Evaluation Criteria in Solid Tumors) complete response, partial response or SD for ≥12 weeks. It was concluded that oral continuous rucaparib dosing is needed for getting better results. Furthermore, the ARIEL2 clinical trial studied the efficacy of rucaparib in 3 groups with relapsed EOC: patients with a BRCA mutation, patients with high loss of heterozygosity (LOH) as a definition for HDR deficiency, and patients with low LOH.45 Results demonstrated a median PFS of 12.8, 5.7, and 5.2 months, respectively. PFS was found to be considerably on higher side in the BRCAm (HR: 0.27 p < 0.001) and LOH high group (HR: 0.62, p = 0.011) as compared with the LOH low group.
Combination Therapy
PARPi and Chemotherapy
Olaparib with Chemotherapy
To achieve synergistic action and further improve clinical efficiency, PARPis have been recommended in combination with chemotherapy. However, this combination is challenging because of overlapping bone marrow toxicity.49 Phase I clinical trial was conducted to further study and decide the safety of olaparib with cisplatin in patients with advanced breast cancer, other solid tumors, and EOC. The MTD could not be established as none of the cohorts reached dose-limiting toxicity levels and, therefore, it was finally concluded that the scheme of cisplatin 60 mg/m2 (day 1, q21 days) in combination therapy with intermittent 50 mg olaparib capsules (days 1–5) was a tolerable dose. Consequently, Oza and team50 investigated, in a randomized phase II clinical trial, the combined therapy of olaparib (200 mg BID) with carboplatin (AUC4) and paclitaxel (175 mg/m2) followed by olaparib maintenance therapy, as compared with standard carboplatin (AUC6) therapy and paclitaxel (175 mg/m2) in patients with platinum-sensitive, recurrent, high-grade serous EOC. The PFS was found to be 12.2 months (95% CI, 9.7–15.0) against 9.6 months (95% CI, 9.1–9.7) in favor of the olaparib arm (HR 0.51; p = 0.0012). In patients with BRCA mutations, this difference was found to be even greater (HR 0.21; p = 0.0015). Finally, it was concluded that the combination cohort had an acceptable and manageable tolerability profile but it required upfront dose reduction of chemotherapy. Dent51 investigated the tolerability and toxicity of olaparib in combination with weekly paclitaxel (90 mg/m2) in 19 patients with metastatic triple negative breast cancer. Preliminary data did not reach a MTD.
Veliparib with Chemotherapy
Veliparib is also researched in combination with chemotherapy. For EOC, Bell-McGuinn52 compared 3 arms of veliparib plus chemotherapy and bevacizumab in a phase I clinical trial in patients with previously untreated EOC. The first arm was given veliparib plus carboplatin (AUC6), paclitaxel (175 mg/m2), and bevacizumab (15 mg/kg); the second arm was administered with veliparib plus carboplatin, a lower dose of paclitaxel (80 mg/m2), and bevacizumab; and the third arm received veliparib plus cisplatin (75 mg/m2), paclitaxel (60 mg/m2), and bevacizumab. Primary results demonstrated a RP2D of veliparib of 150 mg BID for all schedules. Veliparib has also been researched in combination with cyclophosphamide with BRCAm or high-grade serous EOC.53 This combination did not result in an improved ORR (partial response [PR]: 11.8 vs. 19.4%; complete response [CR]: 2.9 vs. 2.7%) or PFS (2.3 vs. 2.1 months; p = 0.68) as compared with cyclophosphamide alone. Gray and team54 focused on the effects of veliparib plus carboplatin and gemcitabine in advanced solid tumors in a phase I clinical trial. Results demonstrated a RP2D of 250 mg veliparib with carboplatin (AUC 4) and gemcitabine (800 mg/m2) and responses were reported in 69% of the patients with BRCAm EOC. The efficacy of veliparib with carboplatin and paclitaxel is currently under further investigations. NCT02470585 is a phase III clinical trial by the Gynaecologic Oncology Group (GOG), which is currently researching veliparib with carboplatin and paclitaxel followed by maintenance therapy in patients with primary EOC.55
For breast cancer, Loibl et al56 researched the combination therapy of veliparib (50 mg), carboplatin (AUC6) and paclitaxel (80 mg/m2) for stage II–III triple negative early breast cancer as neoadjuvant therapy in the BrighTNess trial, in which three arms were compared: veliparib, carboplatin, and paclitaxel versus carboplatin and paclitaxel versus paclitaxel. BRCA-1 or BRCA-2 mutations were found in 14% of the veliparib, carboplatin, and paclitaxel group; 16% in the carboplatin and paclitaxel group; and in 15% of the paclitaxel group. Results demonstrated a considerably higher pathological complete response for veliparib, carboplatin, and paclitaxel group in comparison with paclitaxel alone (53 vs. 31%; p < 0.0001), but a similar pathological complete response in comparison with the carboplatin plus paclitaxel group (53 vs. 58%; p = 0.36). A phase I clinical trial by Rodler30 researched the combination of veliparib (300 mg BID) with cisplatin (75 mg/m2) and vinorelbine (25 mg/m2) in patients with advanced triple negative and/or BRCAm breast cancer. The combination showed an ORR of 35% (95% CI 23–50) with a tolerable safety profile. At present investigations on the efficiency of veliparib plus temozolomide57 and of veliparib plus carboplatin in patients with breast cancer4 are ongoing.
Niraparib with Chemotherapy
A study researching niraparib in combination with carboplatin, carboplatin, and paclitaxel or carboplatin and pegylated liposomal doxorubicin (NCT0111060358) and another study with niraparib in combination with pegylated liposomal doxorubicin (PLD) (NCT0122794159) in solid tumors and EOC have been stopped without any further explanation.
Rucaparib with Chemotherapy
Plummer60 first investigated pharmacodynamics and pharmacokinetics of rucaparib in combination with temozolomide in patients with advanced solid tumors. Results demonstrated a PARP inhibitory dose of 12 mg/m2. At present, results are awaited for phase II/III clinical trials of PARPis in combination with chemotherapy.
PARPis and Immunotherapy
There are numerous immune checkpoint inhibitors; pembrolizumab and nivolumab target the programmed death protein-1 (PD-1), avelumab, atezolizumab, and durvalumab target the programmed death ligand-1 (PD-L1) and ipilimumab and tremelimumab target cytotoxic T-lymphocyte–associated protein 4 (CTLA-4). These immune checkpoint inhibitors have been found to be beneficial in treating several types of malignancies and are currently under evaluation in female cancer patients. The combination of PARP inhibition and immune checkpoint inhibition showed promising results in patients with EOC and HDR deficiency.61 Tumors with a BRCAm typically harbor TP53 mutations and have a higher mutational load.54 Hence, they also harbor a greater number of neoantigens that enhance the recruitment of tumor infiltrating lymphocytes (TILs); in BRCAm tumors a considerably increased expression of CD3+ and CD8+ TILs have been demonstrated,55 as well as increased expression of PD-1 and PD-L1 as compared with wild type EOC.62 Furthermore, PARP inhibition can modulate immune signaling pathways through various mechanisms,63 both activating and nonactivating. In vitro a CTLA-4 antibody, but not PD-1/PD-L1 blockade, synergized therapeutically with veliparib.64 While the PARPi talazoparib increased the number of peritoneal CD8+ T-cells and natural killer cells and increased production of interferon (IFN)-γ and tumor necrosis factor-α in a BRCA1-mutated OC xenograft model.65 The exact immune-modulating effects of checkpoint inhibitor plus PARPi combinations are currently under investigations and needs further research. Currently, multiple trials are, therefore, investigating these combinations.66
Moreover, it is still unclear in which clinical setting the use of immune checkpoint inhibitors in EOC is most favorable. PARPis in combination with immune checkpoint inhibitors might be most beneficial in primary disease or early recurrence due to a lower tumor burden. Future clinical trials will test these combinations in first-line treatment of breast and EOC.
Antiangiogenic Therapy
Preclinical and clinical evidences suggest that there are existing interactions between the VEGF pathway and PARP inhibition. Multiple groups have demonstrated that PARP inhibition reduces VEGF-induced angiogenesis. A preclinical study conducted by Bindra and team67 demonstrated that hypoxia is related with impaired HDR and, therefore, a state of BRCAness. They also postulated that in a hypoxic state the cells are pushed toward NHEJ because of impaired HDR and thus show increased genetic instability and cell death. Liu et al68 designed a phase I clinical trial with olaparib bid and cediranib, a tyrosine kinase inhibitor directed against VEGF. The RP2D was cediranib 30 mg daily with olaparib 200 mg bid. Subsequently, Liu69 investigated the efficiency of olaparib in combination with cediranib in a phase II clinical trial in 90 patients with recurrent platinum-sensitive EOC. Results demonstrated a PFS of 17.7 months for treatment with olaparib and cediranib against 9.0 months for olaparib monotherapy. A post hoc analysis demonstrated that median PFS was even greater for patients with a g BRCA-1/2m: 19.4 months in the combination arm as compared with 16.5 months in the olaparib monotherapy group, respectively. Most common grade-3 toxicities reported in the olaparib plus cediranib group were fatigue, hypertension, and diarrhea. Dose reductions were necessary in 77 and 24% of patients, respectively. Zimmer70 conducted a phase I clinical trial investigating the RP2D for durvalumab + olaparib + cediranib in recurrent female cancers. They concluded a RP2D of 1,500 mg q28d durvalumab + 300 mg BID olaparib + 20 mg 5 days on/2 days off cediranib is tolerable and active. Overall, antiangiogenetic therapy in combination with PARP inhibition showed promising results due to its efficacy and potential synergism. At present many clinical trials are investigating the combination therapy of bevacizumab or cediranib with PARPis in ovarian or breast cancer.
Resistance to PARPis
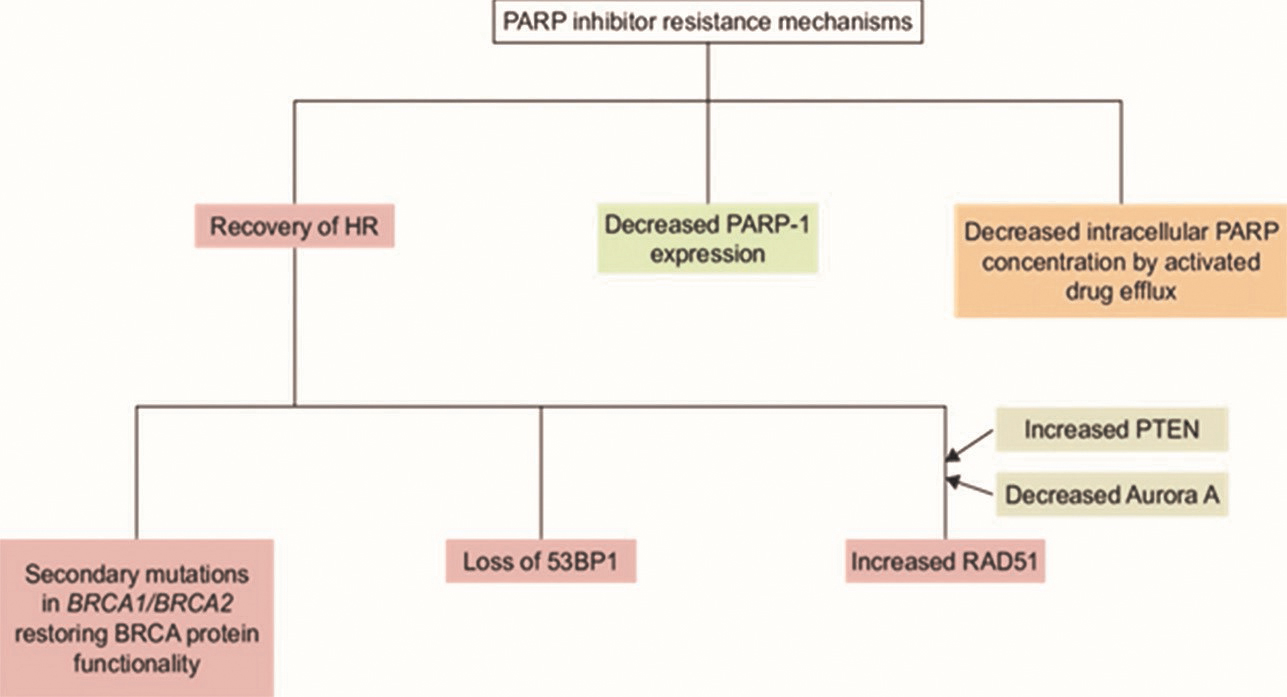
Lord and Ashworth described four pathways leading to resistance to PARPis (Fig. 3).71 Most of the clinical studies conducted in vitro, with mice with different kind of knockout genes. The first mechanism is the incidence of a secondary mutation in the affected BRCA gene that would lead to the restoration of the BRCA open reading frame. Due to this restoration, the BRCA gene can be translated and it can further lead to (partial) functional protein to repair DSBs. Multiple studies have found this phenomenon in patients who had developed resistance to PARPis. The second mechanism depends on the (partial) restoration of HDR due to the somatic loss of expression of genes involved in the regulation of DSB repair pathway choice, like the tumor suppressor p53-binding protein 1 (53BP1) or REV7.72 This mechanism is shown in vivo in mice. A very recent publication describes the identification of shield in, a complex of REV7, RINN1, RINN2, and RINN3 proteins. This complex restrains DNA end resection and thereby promotes NHEJ. Deletion of one of the shield in components leads to resistance to PARPis in BRCA-1 depleted cells.73 Third, the upregulation of the P-glycoprotein efflux pump, pumps PARPis out of the cell, resulting in a decreased inhibition of PARP. The fourth mechanism is the hypothesis that poses that acquired PARP1 loss-of-function mutations or down-regulation of transcription can result in PARPi resistance.
-
Fig. 3 PARP inhibitor resistance mechanisms. BRCA, breast-related cancer antigens; PARP, poly (ADP-ribose) polymerase; PTEN, phosphatase and tensin homolog.
Another mechanism explaining resistance to PARPis is replication fork stabilization. Deficiencies in PAX-interacting protein 1(PTIP), chromodomain-helicase-DNA-binding protein 4 (CHD4), and PARP-1 limit the action of MRE11 to single-strand DNA at stalled replication forks. MRE11 is involved in the degradation of stalled replication forks. When MRE11-dependent replication fork degradation is absent due to deficiencies in PTIP, CHD4, or PARP1, nascent DNA strands will be protected from degradation; therefore, the cell will be resistant to PARP inhibition.74 BRCA-deficient cells become resistant to different types of DNA-damaging agents through the loss of PTIP, PARP1, and CHD4. Furthermore, survival analysis of patients with EOC with a BRCA2 mutation treated with platinum chemotherapy demonstrated that high PTIP expression has a correlation with a longer PFS.
Furthermore, increased phosphorylation of ribosomal protein S6 leading to upregulation of the mTOR pathway75 and upregulation of NF-kB (nuclear factor kappa light chain enhancer of activated B cells) signaling can also lead to PARPi resistance. Depending on these mechanisms it was hypothesized that PARPi-resistant tumors should be treated with rapamycin (an mTOR [mammalian target of rapamycin] inhibitor) or with bortezomib (a proteasome inhibitor). The combination of PARP inhibition and rapamycin effectively suppressed tumor growth in mice69 and the combination of PARP inhibition plus bortezomib led to increased cell death in PARPi–resistant cells.76 Further investigations (both preclinical and clinical) of mechanisms of PARPi resistance will direct us to strategies that will optimally use PARPis in the clinic.
Biomarkers
As far as the application of PARPis is considered beyond BRCAm tumors, it is necessary to identify those classes of patients that get benefited mostly from PARPis to maximize the effects of treatment, further prevent futile therapy and toxicity, and limit the financial burden of health care. Multiple studies have already demonstrated that there is an additional group of tumors with HDR deficiency that also respond to PARPis.59 These tumors have a so called “BRCAness” phenotype, a deficiency in HR in the absence of a BRCA mutation, which makes them responsive to PARPis.
At present, different approaches have been implemented to further identify HDR deficiency or BRCAness. The first way is to analyze tumors for loss of functional mutations in genes that are involved in HDR. These genes include ataxia telangiectasia mutated (ATM), ataxia telangiectasia and Rad3-related (ATR), CHEK2, BRIP1, PALB2, RAD51C, and RAD51D. The second way is to detect BRCAness is via transcriptional signatures. Larsen and team77 analyzed 55 familial germ line BRCA-1 or BRCA-2 mutated breast cancer patients and 128 patients with sporadic breast cancer. They designed a transcriptional signature to distinguish BRCA-1 tumors from sporadic tumors with an accuracy of 83% and BRCA-2 tumors with 89%, which was validated in independent datasets. This type of transcriptional signature might also be implemented to identify BRCAness in non BRCA-related tumors. The third way to detect BRCAness is through the detection of a genomic signature in a tumor. These signatures represent a pattern of mutations or genomic alterations which are characteristic for the use of error-prone repair pathways in the absence of HDR.78 These patterns consist of specific nucleotide substitutions (e.g., mutational signature 3) or sequence microhomology at breakpoints.79 An algorithm dependent on the integration of six signatures associated with BRCA deficiency (including somatic nucleotide substitutions, insertion/deletion, and rearrangement patterns), termed HRDetect was developed by Davies.80 Nowadays, SNP-based profiling has also been implemented to define a so-called HDR deficiency score, based on the combination of 3 DNA-based measures of genomic instability (i.e., loss of heterozygosity [LOH], telomeric allelic imbalance [TAI], and large-scale transitions [LST]).81 This is the myChoice HDR deficiency test (Myriad Genetic) which was used in the NOVA clinical trial as discussed earlier, but it could not discriminate between patients that demonstrated a benefit from treatment niraparib or not.82 A more recent approach to detect BRCAness is through functional biomarkers or a functional test for current HDR deficiency.59 After the induction of DNA DSBs in fresh tumor tissue ex-vivo, RAD51 protein will accumulate at the sites of the breaks. This key step in the HDR pathway can be visualized by immunofluorescent staining as foci in the nucleus. The inability to form RAD51 foci after the induction of DSBs in replicating tumor cells is a biomarker for BRCAness. This biomarker technique requires fresh tumor tissue obtained before starting chemotherapy.83 Finally, in the ARIEL3 clinical trial,48 the percentage of genome-wide LOH quantification was used to compare effectiveness of rucaparib among groups with high or low genomic LOH levels. Tumors with high LOH appear to respond better to PARPi therapy and, therefore, genomic LOH quantification might be used to identify patients who might benefit from PARPis.84
PARPis and Antiangiogenic Agents
Overexpression of PARP-1 has been noted to exert a clear proangiogenic effect in EOC by upregulation of VEGFA.85 Antiangiogenic therapies are well known to induce a hypoxic cellular state, leading to down regulation of HR repair genes (BRCA1, BRCA2, and RAD51), with consequent enhancement of PARPis sensitivity.86 Hypoxia is also found to be related to hypoxia inducible factor–1 α (HIF1α) upregulation, which is considered one of the most common mechanisms of resistance to angiogenesis inhibitors. Interestingly, PARP-1 may play a pivotal role in HIF-1α stabilization.85 As a consequence, the use of PARPis may prevent its accumulation and signaling, thus overcoming this resistance mechanism, resulting in death of the targeted hypoxic cell. This is a suitable example of “contextual synthetic lethality” because of tumor microenvironment, in which hypoxia-induced repair-deficient tumor cells can be targeted by disrupting backup pathways. The combination of cediranib, an oral tyrosine kinase inhibitor of VEGF receptor (VEGFR) and olaparib versus olaparib alone was tested in a phase II clinical trial in platinum-sensitive ROC.51 Median PFS was considerably longer for cediranib/olaparib-treated patients than for patients on olaparib alone (17.7 vs. 9 months, HR 0.42; p = 0.005). A post hoc exploratory analysis showed an increased activity of cediranib plus olaparib versus olaparib alone in the subgroup of patients with wild type or unknown BRCA status, with an improvement in the median PFS from 5.7 to 16.5 months (HR = 0.32; p = 0.008) and in the ORR from 32 to 76% (p = 0.006). Among gBRCAm patients, there was a lesser trend toward increased activity for the combination arm, with a slighter gain of PFS (16.5–19.4 months) and ORR (benefit 63–84%). Notably, side effects (fatigue, diarrhea, and hypertension) of any grade occurred in the combination arm, resulting in dose reductions in more than 75% of patients. Three phase III clinical trials are currently ongoing to validate this combination in different settings. The aim of GY004 trial was to compare olaparib monotherapy versus olaparib and cediranib combination versus standard platinum-based chemotherapy in patients with platinum-sensitive recurrent ovarian/fallopian tube cancers. GY005 trials are evaluating the same therapeutic options in recurrent platinum-resistant or refractory OC. Finally, ICON 9 trial is examining maintenance therapy with cediranib and olaparib or maintenance olaparib alone after platinum-based chemotherapy in patients with recurrent platinum-sensitive high-grade OC. The combination of olaparib 400 mg BID with bevacizumab 10 mg/kg q2w was investigated in a phase I study in patients with advanced solid tumors; a good tolerability without serious AEs or dose-limiting toxicities were recorded.87 On the basis of these results, a phase III trial, PAOLA1, was planned to determine the efficacy of olaparib or placebo combined with bevacizumab as maintenance treatment in patients with OC treated with standard first-line platinum-based chemotherapy plus bevacizumab. Finally, the results of a phase I clinical trial of bevacizumab (15 mg/kg q21 days) plus niraparib (300 mg oral QD) demonstrated an ORR of 45% in 12 patients and DCR of 91% in the combination treatment arm.53 Class toxicities (hypertension, anemia, thrombocytopenia, fatigue, constipation, and nausea) were manageable and only one dose-limiting toxicity was reported: grade 3 thrombocytopenia that persisted for ≥5 days. Results from AVANOVA, a phase II ongoing clinical trial, comparing single-agent niraparib with niraparib plus bevacizumab in 94 women with platinum-sensitive OC, are still pending.
PARPis and Immune Checkpoint Inhibitors
Achievement of tumor antigenicity sensitizes cancers to checkpoint blockade therapy. Multiple clinical studies have demonstrated that BRCA-1/2m and wt-BRCA1–2 HRD ovarian tumors display a higher neoantigen load than HR-proficient cancers.54 PARPis have been considered to enhance the response to immunotherapy in HRD OC by yielding a greater mutational burden, thereby expanding neoantigen expression. In fact, the presence of danger signals, followed by DNA damage, activates the stimulator of interferon genes (STING) pathway which plays a vital role in the innate immunity by inducing type I interferon and proinflammatory cytokine production.55 Moreover, PARPis administration upregulates PD-L1 expression on tumor cells, which in turn attenuates PARPis efficacy via cancer-associated immunosuppression. As a consequence, the targeted blockade of PD-L1 pathway can restore antitumor immunity and potentiate the antitumor activity of PARPis.56 Altogether, these observations provide a scientific rationale for evaluating the combination of immune checkpoint blockade with PARPis in OC clinical trials. The phase I/II TOPACIO clinical trial3 demonstrated niraparib in combination with pembrolizumab, an anti–PD-1 antibody, to be a promising therapeutic option for the management of platinum-resistant OC. Following dose finding in phase I, the RP2D of niraparib and pembrolizumab was assessed at 200 mg orally QD and 200 mg IV every 21 days, respectively. During phase II, among 60 out of 62 enrolled patients who were considered evaluable for initial response assessment, 64% of them were platinum-resistant, 19% had platinum-refractory disease, and 17% had platinum-sensitive OC ineligible for further platinum. In the whole population, ORR and DCR were 25 and 68%, respectively, while in the BRCA1/2m cohort, ORR and DCR were 45 and 73%, respectively.
In regard to olaparib, phase I/II basket MEDIOLA clinical trial evaluated the combination with durvalumab, an anti-PDL1 antibody, for the management of gBRCA 1/2m platinum-sensitive relapsed OC.57 Thirty-two patients were given olaparib 300 mg (tablets) BID for the period of 4 weeks, followed by a combination of olaparib 300 mg BID and durvalumab 1.5 g intravenous (IV) every 4 weeks, until disease progression. DCR at 12 weeks was 81% while ORR was 63%. This combination was well tolerated, with a low incidence of grade 3 toxicities and all-grade immune-related AEs. The results of TOPACIO clinical trial and MEDIOLA study were presented at European Society of Gynaecological Oncology (ESGO) congress 2018. In another phase II clinical trial the combination of durvalumab and olaparib was tested in 34 ROC patients with platinum-sensitive or -resistant disease. The preliminary efficacy results, which were presented during the ESMO congress 2018, showed a response rate and DCR of 15 and 53%, respectively. The combination of olaparib at a dose of 300 mg BID with tremelimumab, a CTLA-4 antibody, at a dose of 10 mg/kg monthly, was tested in another phase I clinical trial4 for the treatment of BRCA-1/2m ROC and demonstrated good safety.
Multiple clinical studies assessing the efficacy of PARPis in combination with immunotherapy are ongoing. A phase I/II trial is evaluating side effects and optimal dose of olaparib in combination with durvalumab and tremelimumab for the treatment of ROC. The phase III FIRST clinical trial was designed to evaluate platinum and TSR-042 (PD-L1 inhibitor) followed by niraparib and TSR-042 maintenance therapy versus adaptive standard platinum-based treatment for newly diagnosed advanced OC patients. In addition, ENGOT-ov46/AGO/DUO-O is a phase III clinical trial whose aim is to assess the efficacy and safety of standard of care platinum-based chemotherapy and bevacizumab followed by maintenance bevacizumab either as monotherapy, or in combination with durvalumab, or in combination with durvalumab and olaparib for newly diagnosed advanced OC patients. Regarding rucaparib, phase III ATHENA study is investigating the association with nivolumab as maintenance therapy in OC patients, after response to frontline platinum-based chemotherapy.
PARPis and Other Therapeutic Agents
As PARPis emerged as an efficient therapeutic strategy in the management of HR-deficient tumors, the real question is to establish whether PARPis can also be effective in the treatment of HR-proficient carcinomas. The condition of HR proficiency, which plays a major role in PARPis resistance, can occur de novo or be acquired. In the latter case, epigenetic or genetic events occurring under selective pressure due to PARPis exposure are responsible for reversing the original HR alterations, hence leading to HR restoration. The most commonly acquired mechanism of resistance to PARPis is a somatic genetic reversion of the original truncating BRCA mutation that restores functional protein expression.58 Alternatively, an acquired epigenetic reversion of BRCA-1, like promoter hypermethylation, has been noted to restore normal BRCA-1 protein expression levels.88 Of note, tumors carrying a specific BRCA-1 mutation, which further disrupts the N-terminal RING domain, respond poorly to platinum drugs and PARPis and rapidly develop resistance.59 Another possible mechanism of PARPis resistance may be the decreased expression of PARP enzymes due to epigenetic silencing or accelerated/high protein turnover.60 Furthermore, intracellular concentrations of PARPis could be reduced by increased P-glycoprotein–mediated efflux, thus resulting in decreased antitumor effect.61 The loss of 53BP1 in BRCA1m tumors, partially restoring the error-free repair mechanism mediated by HR, enhances DNA-damage tolerance and induces PARPis resistance.54
Finally, HR proficient OC with concurrent amplification of cyclin-E genes has been demonstrated to be resistant to PARPis.55 As a consequence, the rationale to combine PARPis with molecularly targeted agents capable of inhibiting HRR may represent a promising effective strategy to expand their use in HR-proficient OC.84 Among all the combinations that are currently under assessment, the association with PI3K inhibitors appears the most reasonable approach, as its phase I evaluation in ovarian and triple-negative breast cancers has just been completed.62 The rationale for this trial was based on two preclinical studies published in 2012,63 which showed that PI3K inhibition significantly decreased BRCA1/2 expression, thus resulting in acquired HRD underlying the antitumor effects of PARPis. In the phase I study, combined exposure to olaparib and BMK120 showed an ORR of 29% in 46 advanced OC patients, irrespective of platinum-sensitivity status.64 The maximum tolerated dose was 50 mg QD of BKM120 and 300 mg BID of olaparib. Randomized phase II studies are warranted to further define the efficacy of PI3K/PARP-inhibitor combinations compared with PARPis alone in different settings of ROC.66 Finally, the use of PARPis after PARPis represents an interesting field of investigation. In this direction, the OReO/ENGOTOv-38 is ongoing trial that will evaluate efficacy and safety of maintenance retreatment with olaparib in patients that retain platinum sensitivity despite progression on olaparib maintenance therapy.
PARPis (Olaparib) Maintenance Therapy in Platinum-Sensitive Relapsed OC
Olaparib, a PARP inhibitor, has formerly revealed efficiency in a phase 2 study when administered in capsule formulation to all-comer patients with platinum-sensitive, relapsed high-grade serous ovarian cancer (HGSOC). Lauraine et al46 in 2017 noted these findings in patients with a BRCA1 or BRCA2 (BRCA1/2) mutation with the help of a tablet formulation of olaparib. Olaparib maintenance therapy provided a noteworthy progression-free survival enhancement with no unfavorable consequence on quality of life in patients with platinum-sensitive, relapsed OC and a BRCA1/2 mutation. Except from anemia, toxicities with olaparib were inferior and manageable. Ledermann et al in 2012 studied olaparib maintenance therapy in platinum-sensitive relapsed OC. They found that olaparib as maintenance therapy considerably improved progression-free survival among patients with platinum-sensitive, relapsed, HGSOC. Interim analysis showed no overall survival benefit. The toxicity profile of olaparib in their study population was consistent with previously conducted studies.89
Treatment of Platinum Refractory OC
The chronological and current keystone of treatment for advanced OC is prime debulking surgery followed by subsequent platinum-based chemotherapy. Regrettably, the soaring initial response rates gained with platinum therapy are not long lasting, as chemo sensitivity typically gives way to a broad chemo cross-resistance that encompasses not just platinum but also multiple drugs to which patients were never previously exposed.90 The dismal 5-year survival rate of 17%91 in stage 4 EOC is mainly attributable to the materialization of resistance, which has a genetic and epigenetic origin.92 However, in contrast to the permanence of genetic defects, epigenetic mechanisms are forceful and potentially reversible with epigenetic therapies, which accounts for their appeal when used as pretreatment “primers” to prevent or reverse nonresponsiveness to chemotherapy. Previous experience with RRx-001, an investigational tumor-related macrophage and neutrophil depolarizing agent with epigenetic and vascular normalization properties, has demonstrated resensitization to platinum in other tumor types, like colorectal, small cell lung cancer (SCLC), and non-SCLC (NSCLC).93
Upfront Role of Olaparib
Olaparib maintenance therapy which is mostly introduced orally at the dose of 400 mg was as efficient and well tolerated as other therapies with admiration to effectiveness mainly calculated by PFS, OS, and quality of life and adverse events. High-quality evidence was noted that women with diverse types of OCs who received olaparib had noteworthy improvements in PFS. Other RCTs about OC patients with olaparib treatment are still ongoing.94
It was worth mentioning that a randomized trial110 compared olaparib doses and reported that the 400 mg dose was superior to the 200 mg dose for median PFS times. So, 400 mg can be considered as a recommended dose. Analyzing results showed that olaparib maintenance therapy led to a considerably longer PFS and a slightly better OS in OC patients. With the help of FACT-O questionnaires for the health-related quality of life, there were no considerable differences in development or deterioration rates between the olaparib group and other interventional groups. In the incorporated studies, the acceptability profile of olaparib treatment was consistent with that reported previously.43 As compared with placebo, olaparib caused the most general adverse events counting diarrhea, nausea, vomiting, anemia, and fatigue at the doses of 300 or 400 mg twice per day. Nevertheless, as compared with other chemotherapy drugs, olaparib could not increase the incidences of the majority of adverse events. Hence it was confirmed that olaparib continuation therapy was generally successful and well tolerated in patients with BRCA-mutated advanced OC.95
Resistance to PARPis
Underlying mechanisms of resistance to PARP inhibition consist of the development of secondary mutations, deficiencies in the NHEJ pathway, and the loss of 53BP1 expression, which lead to an increase in drug export and decreases in PARP1 expression.96 BRCA1/2 mutations remain as a strongest genetic indicator of sensitivity to PARPis.97 However, 40 to 70% of BRCA1/2-mutated OCs not succeed to act in response to PARPis.50 Additionally, the outstanding efficacy of PARPis in OC is not limited to patients with germ line BRCA1/2 mutations but extends to those with tumors with HR repair pathway deficiencies. Tumors with mutations in certain genes that are not directly concerned in DNA repair but are related to the HR pathway or influence the effect of HR pathway genes are responsive to PARPis.98
The most familiar mechanism acquired of resistance to PARP inhibition is the reinstatement of BRCA1 or BRCA2 protein function by secondary mutations. Additionally, this mechanism is shared by platinum resistance and PARPi resistance. In vitro selection of a BRCA2-mutated OC cell line, which was susceptible to both platinum and PARP inhibition, by a cisplatin/PARP inhibitor combination led to the recovery of BRCA2 function induced by secondary BRCA2 mutation. OC cells extracted from ascites of platinum-resistant relapsed OCs were found to harbor secondary BRCA2 mutations and to be BRCA2-proficient. The depletion of BRCA2 resensitized these cells to the cisplatin/PARPi combination.99 Additionally, the secondary mutations in RAD51C or RAD51D were found to be associated with resistance to PARP inhibition.
Sequencing of two samples of olaparib-resistant tumors from clinical trials found secondary BRCA2 mutations that restored the ORF of the gene.100 On the other hand, in another study, deep sequencing of six olaparib-resistant tumors showed no evidence of secondary BRCA mutation.101 BRCA1/2-mutated OCs harboring secondary mutations and exhibiting progression following platinum treatment may be found to be resistant to both platinum and PARPis. However, platinum-resistant, BRCA1/2-mutated OCs without secondary mutations are likely to be sensitive to PARPis. Thus, testing secondary mutations may notify treatment options for OC patients.102 For example, 6-thioguanine has been demonstrated to kill cells and tumors that have gained resistance to PARPis via the genetic reversion of the BRCA2 gene.103 Inhibition of PARP enzymes by PARPis would reduce the combined action of these NHEJ proteins and poly (ADP-ribose) polymers, thus promoting NHEJ and genomic unsteadiness.104 Functional studies demonstrated that NHEJ deficiency was independent of HR proficiency and was found to be associated with resistance to PARPis in ex vivo primary cultures.
PARP1 is a nuclear enzyme that is activated by DNA damage and plays a significant role in BER. The inhibition of PARP1 is not equivalent of PARP1 deletion and the mechanisms of action for PARPis are dependent on both the catalytic inhibition of PARP1 enzyme and the trapping of PARP1-DNA complexes. In vitro studies have demonstrated that the PARP inhibitor olaparib has no effect on cells with complete absence of the PARP1 enzyme. In the presence of PARPis, dysfunctional PARP1 enzymes bind DNA and form PARP1–DNA complexes and PARPis promote trapping of these PARP1–DNA complexes that are toxic to the cell.23 A certain amount of functional PARP1 is critical to tumor responses to PARPis as PARP1 is obligatory both as a substrate for PARP1 trapping and for the cytotoxicity of PARPis. The deletion of PARP1 has been verified to cause resistance to all PARP inhibitors in OC cell lines in vitro.105 The PARP1 expression level is positively correlated with PARP inhibitor sensitivity.106 For example, an acquired short expression level of PARP1 is a potential cause of resistance to PARP inhibitors in PDX models.105 Furthermore, cells with PARP1 mutations were 100-fold more resistant to PARPis than were cells with wild-type PARP1.107 Mutations both within and outside the PARP1 DNA-binding domains modify PARP1 trapping and induce PARP inhibitor resistance.
Finally, to conclude about resistance to PARPis homologous recombination deficiency, which can be investigated through the mutational analysis of HRD gene panels, genomic scar analysis and functional assays, remains a strong predictor of clinical benefit from PARPis. However, the HRD biomarker apparently cannot competently identify the subgroup of patients with wild-type BRCA that will achieve a considerable increase in PFS with PARP inhibitor treatment. This inability may result from other HRD mechanisms not detected by current assays or from alternative explanations for PARP inhibitor sensitivity. In addition, numerous combination treatment strategies can induce HR pathway deficiency. Notably, the response to platinum-based chemotherapy remains a strong predictor of the response to PARP inhibitor therapy, especially for BRCA-proficient OC. Mechanisms of resistance to PARP inhibition consists of the secondary developmental mutations, deficiencies in the NHEJ pathway, the loss of 53BP1 expression, and increases in drug export and decreases in PARP1 expression. The importance of these mechanisms of PARP inhibitor resistance in clinical settings and the identification of strategies to overcome this resistance warrant further investigation.
Related Clinical Trials of PARPis
Clinical studies of PARPis, including olaparib, rucaparib, and niraparib, in OC are summarized in Table 2. To begin with, the FDA permitted olaparib as the fourth-line treatment for advanced OC with gBRCAm, based on the results from a phase II study representing an objective response rate (ORR) of 31% and a median overall survival (OS) time of 16.6 months with olaparib treatment in 193 OC patients [NCT01078662]. Patients with platinum-resistant disease or those unsuited for further platinum therapy due to noteworthy toxicity or hypersensitivity to platinum, were also included in this study. This level of activity appreciably exceeded that of conventional third-/fourth-line therapy; hence, the FDA approved olaparib for this indication.108 A pooled analysis of 6 phase I/II clinical trials [NCT00516373 (Study 2), NCT00777582 (Study 24), NCT00494442 (Study 9), NCT00628251 (Study 12), NCT00679783 (Study 20), and NCT01078662 (Study 42)] recognized the ORR as 36% and the median duration of response as 7.4 months with olaparib treatment among patients with gBRCAm and advanced relapsed OC. The ORR among patients who had received three or more lines of prior chemotherapy was found to be 31%, with duration of response of 7.8 months, showing that a sustained response to olaparib could be achieved in heavily pretreated, relapsed, gBRCAm-associated OCs.109 The highest tolerated dose (MTD) of olaparib was recognized as 400 mg twice daily in a phase I trial [NCT00516373].44
|
Olaparib |
||||
|---|---|---|---|---|
|
NCT Number |
Phase |
Clinical Trial |
||
|
1. |
NCT02282020 SOLO3 |
III |
Randomized |
Olaparib tablets 300 mg po bid vs. physician choice single-agent nonplatinum-based chemo for gBRCAm platinum-sensitive relapsed HGS/EOC following ≥2 platinum-based chemo with progression ≥6 months after last platinum |
|
2. |
NCT02446600 |
III |
Randomized |
Platinum-based chemo (carboplatin + paclitaxel; carboplatin + gemcitabine; carboplatin + PLD) vs. olaparib vs. olaparib + cediranib for platinum-sensitive relapsed HGS/EOC or gBRCAm HGOC with any number of platinum-based chemo and ≤1 nonplatinum therapy with CR to last platinum |
|
3. |
NCT02502266 |
II/III |
Randomized |
Physician choice chemo (paclitaxel; PLD; topotecan) vs. olaparib + cediranib vs. olaparib vs. cediranib for platinum-resistant or refractory relapsed, HGS/EOC non-gBRCAm or HGOC gBRCAm with ≤3 prior regimens and ≤1 nonplatinum |
|
4. |
NCT02889900CONCERTO |
IIb |
Nonrandomized |
Noncomparative cediranib + olaparib for recurrent platinum-resistant OC without gBRCAm |
|
5. |
NCT03106987OReO |
IIIb |
Randomized |
Olaparib vs. placebo maintenance retreatment for relapsed nonmucinous EOC, who have had disease progression following maintenance therapy with a PARPi and a CR/PR to subsequent platinum-based chemotherapy |
|
6. |
NCT02855697MOLTO |
I |
Nonrandomized |
Noncomparative multimaintenance olaparib for platinum-sensitive relapsed gBRCAm HGS/EOC with 2 or more courses of maintenance olaparib |
|
7. |
NCT03402841OPINION |
IIIb |
Nonrandomized |
Noncomparative olaparib maintenance for platinum-sensitive relapsed non-gBRCAm HGS/EOC |
|
8. |
NCT02340611 |
II |
Nonrandomized |
Non-comparative cediranib + olaparib after disease progression on olaparib alone in OC |
|
9. |
NCT03278717ICON 9 |
III |
Randomized |
maintenance olaparib + cediranib vs. olaparib alone for relapsed OC with disease progressed more than 6 months after first-line chemotherapy or CR/PR to ≥4 cycles of platinum-based chemotherapy. |
|
10. |
NCT03470805 |
II |
Nonrandomized |
Noncomparative olaparib maintenance after response to trabectedin-PLD in recurrent gBRCAm or sBRCAm HGS/EOC |
|
11. |
NCT03117933OCTOVA |
II |
Randomized |
Olaparib vs. olaparib + cediranib vs. weekly paclitaxel for BRCAm platinum-resistant OC |
|
12. |
NCT03161132ROLANDO |
II |
Nonrandomized |
Noncomparative olaparib + PLD for platinum-resistant advanced OC |
|
13. |
NCT01623349 |
I |
Safety |
To determine the safety of oral PI3kinase inhibitor BKM120 or BYL719 + olaparib for recurrent HGSOC |
|
14. |
NCT01650376 |
Ib |
To determine the MTD of olaparib + weekly carboplatin and paclitaxel in relapsed OC |
|
|
15. |
NCT03314740BAROCCO |
II |
Randomized |
Weekly paclitaxel vs. cediranib-olaparib with continuous schedule vs. cediranib-olaparib with intermittent schedule for platinum refractory or resistant recurrent HGSOC |
|
16. |
NCT02983799 |
II |
Nonrandomized |
Noncomparative olaparib for platinum-sensitive or partially platinum-sensitive, relapsed, HGS/EOC with at least 1 prior line of platinum-based chemotherapy, in gBRCAm, sBRCAm, or HRD subgroups |
|
17. |
NCT02571725 |
I-II |
Nonrandomized |
Noncomparative olaparib and CTLA-4 blockade tremelimumab for BRCAm recurrent OC |
|
18. |
NCT02477644PAOLA-1 |
III |
Randomized |
Olaparib vs. placebo for advanced FIGO stage IIIB–IV HGS/EOC with standard first-line platinum-taxane chemotherapy and bevacizumab concurrent and in maintenance, ≥3 cycles of bevacizumab in combination with the 3 last cycles of platinum-based chemotherapy |
|
19. |
NCT01445418 |
I |
Nonrandomized |
Noncomparative olaparib + carboplatin for gBRCAm and sporadic OC |
|
20. |
NCT03462342CAPRI |
II |
Nonrandomized |
Noncomparative ATR inhibitor AZD6738 + olaparib for recurrent HGSOC (platinum-sensitive or -resistant) |
|
21. |
NCT03579316 |
II |
Randomized |
Noncomparative adavosertib AZD1775 alone or with olaparib for recurrent OC during olaparib progression |
|
22. |
NCT03699449AMBITION |
II |
Randomized |
Olaparib + cediranib, durvalumab + olaparib, durvalumab + chemotherapy, durvalumab + tremelimumab + chemotherapy; a biomarker-driven targeted therapy for HRD platinum-resistant recurrent OC |
|
23. |
NCT02345265 |
II |
Nonrandomized |
Noncomparative olaparib + cediranib for recurrent OC |
|
24. |
NCT02121990 |
I |
Dose-escalation study of IP cisplatin, IV/IP paclitaxel, IV bevacizumab, and oral olaparib for newly diagnosed OC |
|
|
25. |
NCT02489006 NEO |
II |
Randomized |
Randomized, neoadjuvant olaparib for platinum-sensitive recurrent HGSOC prior to surgery and chemotherapy |
|
26. |
NCT02953457 |
I/II |
Nonrandomized |
Noncomparative olaparib together with durvalumab and tremelimumab for gBRCAm recurrent or refractory OC |
|
27. |
NCT02898207 |
I |
Safety |
To determine the safety and best dose of olaparib + HSP90 inhibitor onalespib for recurrent OC |
|
28. |
NCT01116648 |
I/II |
Safety |
To determine the safety and best dose of cediranib + olaparib for recurrent OC |
|
29. |
NCT02208375 |
Ib |
To determine the MTD of olaparib + oral mTORC1/2 inhibitor AZD2014 or AKT inhibitor AZD5363 for recurrent OC |
|
Abbreviations: HGS, high-grade serous; EOC, epithelial ovarian cancer; PLD, pegylated liposomal doxorubicin; PARPi, Poly (ADP-ribose) polymerase inhibitors; OC, ovarian cancer; HGSOC, high-grade serous ovarian cancer; HRD, homologous recombination deficiency; MTD, maximum tolerated dose.
A dose–response association between diverse olaparib dose levels was studied in two phase II clinical trials. In the first phase II study [NCT00628251], the ORR was observed to be higher in the 400 mg olaparib group (31%) than in the 200 mg olaparib group (25%).110 In an additional phase II study [NCT00494442], a variation of 3.9 months in the median PFS was reported in the 400 mg olaparib group as compared with the 100 mg olaparib group, in favor of the 400 mg olaparib group. And the ORRs were 33% and 13% in the 400 mg olaparib and 100 mg olaparib groups, respectively.43
SOLO2 and SOLO1 are all randomized, double-blind, placebo-controlled phase II/III clinical trials of olaparib monotherapy that are highly noteworthy showed that olaparib maintenance monotherapy considerably improved PFS (median 8.4 vs. 4.8 months; HR 0.35; 95% CI 0.25–0.49; p < 0.001) as compared with placebo in patients with platinum-sensitive, recurrent HGSOC who had received two or more prior lines of platinum-based chemotherapy and established a CR/PR to the most recent platinum-based chemotherapy.89 Retrospective germ line and somatic BRCA mutation testing was conducted on all patients with an supplementary 2 years of follow-up. A total of 51% of the HGSOC population showed germ line or somatic BRCA mutation, and patients with or without g/sBRCA mutations both obtained the PFS benefit from olaparib maintenance therapy versus placebo, with a greater PFS benefit in the g/sBRCA1/2-mutated group as compared with the wild-type BRCA group. The first, second and third interim OS analyses from study 19 were conducted after 38, 58, and 77% of patients had died, respectively. The final OS analysis was performed after 210 deaths (79% data maturity), after a median follow-up of 6.5 years.111 Neither of the first112 or second interim OS analyses demonstrated a benefit for olaparib versus placebo for either the BRCA1/2-mutated or BRCA wild-type groups in the overall population.113 The third interim OS analysis and the final OS analysis both demonstrated an OS advantage of olaparib against placebo in all patients and inpatients with BRCA mutations. However, the predefined threshold for statistical significance (p = 0.0095) was not met. In the final OS analysis, 32 patients (24%) had received olaparib maintenance therapy for over 2 years, and 15 (11%) had received olaparib maintenance therapy for over 6 years, which showed the long-term safety and tolerability of olaparib maintenance therapy.114 Therefore, olaparib maintenance appreciably improved PFS in patients with platinum-sensitive recurrent HGSOC treated with two or more previous lines of platinum-based chemotherapy, and patients with a g/sBRCA mutation reported the greatest benefit from olaparib. The analyses for time to first subsequent therapy or death, time to second progression, and time to second subsequent therapy or death reported that the PFS benefit was continued until succeeding treatment and that a long-term benefit was achieved irrespective of BRCA1/2 mutation status. The long follow-up time necessary to obtain sufficient OS data increases the chance that postprogression PARP inhibitor therapy and patient crossover will affect the OS data. When excluding the patients from places where placebo patients were treated with postprogression PARP inhibitors, the OS hazard ratio was found to be extensively improved, demonstrating that in study 19, postprogression PARP inhibitor therapy had a confounding effect on the interim OS analysis for patients with BRCA mutations. SOLO2 [NCT01874353] intended to look into the effectiveness and safety of olaparib in platinum-sensitive, recurrent OC patients with a g/sBRCA1/2 mutation who had received two or more lines of previous chemotherapy and confirmed a CR/PR to the most recent platinum-based chemotherapy. The median PFS was considerably longer with olaparib (19.1 months) than with placebo (5.5 months). The PFS benefit from olaparib maintenance as compared with that from placebo in SOLO2 to a large extent exceeded that was reported in study 19, which is not surprising as SOLO2 included only patients with g/sBRCA1/2-mutated tumors.46 Furthermore, heavily pretreated patients with BRCA1/2-mutated OC whose disease progressed following SOLO 1 was the first clinical trial to further investigate the efficacy of olaparib as a first-line maintenance therapy for primary advanced OCs. Patients who had no evidence of disease at the completion of 2 years stopped receiving the trial intervention. Maintenance olaparib led to an extensive development in the PFS of patients with newly diagnosed advanced OC and BRCA mutation, with a difference of ∼3 years in the median PFS for olaparib as compared with that of placebo. The Kaplan–Meier curves for the olaparib group did not appreciably change after 2 years, which suggests an enduring treatment benefit after treatment stop. The second PFS showed a statistically significant upgrading, suggesting that olaparib did not reduce patients’ ability to benefit from subsequent therapy. Considering the significant PFS advantage in favor of first-line maintenance therapy with PARPis, patients would have to undergo g/sBRCA testing immediately after OC diagnosis and adopt the PARPi first-line maintenance therapy if they were found to be positive for g/sBRCA mutation. Moreover, the PFS benefit from maintenance olaparib as compared with that from placebo in SOLO1 also substantially exceeded that in SOLO2, showing that olaparib is more beneficial to BRCA mutation carriers as a first-line maintenance therapy than as third-line treatment.
Latest FDA Guidelines: Major Updates in Maintenance Therapy
The FDA guidelines make quite a few updates in maintenance therapy in stage 2, 3, and 4 disease: Olaparib is recommended as first-choice of maintenance therapy for patients with BRCA1/2 mutations in absolute clinical remission or partial remission (Fig. 4). The recommendation is category 1 for germ line mutations and category 2B for somatic mutations; O’Malley and team noted that this occurred because there were only few patients with somatic mutations studied. The recommendation applies whether or not the patient was previously treated with bevacizumab.

-
Fig. 4 Summary of FDA-approved PARP inhibitors. BRCA, breast-related cancer antigens; FDA, Food and Drug Administration; PARP, poly (ADP-ribose) polymerase.
The recommendation for olaparib is dependent on results from the SOLO-1 clinical trial, which studied the PFS depending on RECIST criteria and noted that as compared with placebo, median PFS was not reached in the olaparib arm and was 13.8 months in the placebo arm.46 Bevacizumab is also suggested for maintenance therapy post remission for patients with partial or whole responses who received it in primary treatment, or for patients with stable disease. Updates for bevacizumab were based on the GOG-218115 and the ICON7 clinical trials, which O’Malley reviewed. GOG-218 was cited in the June 13, 2018, FDA approval for bevacizumab in combination with paclitaxel or carboplatin, followed by bevacizumab as a single agent, for stage 3 or 4 epithelial ovarian, fallopian tube, or primary peritoneal cancer after initial resection.116
In reviewing the ICON7 study results, O’Malley noted that even though the overall results for did not reach statistical significance, bevacizumab was found to be very effective for the highest-risk patients; published results showed that the estimated median PFS was 10.5 months with standard therapy, versus 15.9 months with bevacizumab (HR 0.68; 95% CI 0.55–0.85; P < 0.001).
Conclusion
Deficient therapeutic strategies tailored especially focusing on biomarkers has always been a significant obstacle in the systemic therapeutic management of advanced-stage OC. Recent developments of PARPis offer a therapeutic approach for population with OC. All the EMA–FDA-sanctioned PARPis, olaparib, rucaparib, and niraparib, are especially characterized by a similar types of efficacy in the maintenance setting for patients with platinum-sensitive OC cases. Till date, the overcoming indications of regulatory agencies do not further support the medical oncologists in the process of selecting the best PARPi to administer. Some different types of considerations may help in personalization of the treatment approach depending on the basis of the clinical and molecular characters of the patient.
Primarily, one very much obvious issue requiring examination is the specific toxicity profile demonstrated by each agent: olaparib is widely known to increase serum levels of creatinine in ∼44% of patients, rucaparib has been related to elevated AST and ALT levels in ∼75% of patients, whereas ∼30% of patients receiving niraparib noted grade 3 thrombocytopenia. Regarding the efficacy data, the lack of phase-III data for olaparib, as compared with rucaparib and niraparib, does not support its clinical application in a nonmutated BRCA population.
However, the HRD-positive population treated with niraparib is different from ROC patients managed with rucaparib as in the ARIEL3 HRD positive cohort included also gBRCA 1/2m patients.
Benefit demonstrated by niraparib in the BRCAwt HRD-positive population, it is becoming increasingly important, in clinical practice, to identify these patients through a validated test. In this regard, myChoice HRD test failed as a negative predictive marker of response, since even HRD negative patients, achieved a modest benefit to niraparib therapy. The current challenge is to confer HR deficiency in HR- proficient tumors, thus sensitizing them to PARPis. This is the rationale behind a growing number of clinical trials exploring combination strategies designed to selectively disrupt HR in cancer cells. Further research studies are needed to better define potential predictors of response beyond BRCA mutations and HRD status, thus resulting in a broader target population of OC patients. Finally, looking at exciting results of olaparib in maintenance setting after platinum-based first line chemotherapy for BRCA 1/2m patients, the future matter will be whether to anticipate the maintenance with olaparib in first-line and eventually repurpose it to the sensitive platinum recurrence.
Conflict of Interest
None declared.
Funding This study has not received any financial support.
References
- Poly (ADP-ribose) polymerase inhibitors: recent advances and future development. J Clin Oncol. 2015;33(12):1397-1406.
- [Google Scholar]
- Phase 2 multicentre trial investigating intermittent and continuous dosing schedules of the poly(ADP-ribose) polymerase inhibitor rucaparib in germline BRCA mutation carriers with advanced ovarian and breast cancer. Br J Cancer. 2016;114(07):723-730.
- [Google Scholar]
- Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390:1949-1961. (10106)
- [Google Scholar]
- Frequency of germline mutations in 25 cancer susceptibility genes in a sequential series of patients with breast cancer. J Clin Oncol. 2016;34(13):1460-1468.
- [Google Scholar]
- BRCA1 and BRCA2: breast/ovarian cancer susceptibility gene products and participants in DNA double-strand break repair. Carcinogenesis. 2010;31(06):961-967.
- [Google Scholar]
- Synthetic lethality—a new direction in cancer-drug development. N Engl J Med. 2009;361(02):189-191.
- [Google Scholar]
- Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917-921. (7035)
- [Google Scholar]
- Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376:235-244. (9737)
- [Google Scholar]
- Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med. 2017;377(06):523-533.
- [Google Scholar]
- Research C for DE. Approved drugs—FDA approves olaparib for germline BRCA-mutated metastatic breast cancer. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm592357.htm Accessed
- [Google Scholar]
- What are the risk factors for ovarian cancer? www.cdc.gov/cancer/ovarian/basic_info/risk_factors.htm Accessed
- [Google Scholar]
- Surveillance, Epidemiology, and End Results Program. Cancer stat facts: ovarian cancer. https://seer.cancer.gov/statfacts/html/ovary.html Accessed
- [Google Scholar]
- Challenging and complex decisions in the management of the BRCA mutation carrier. J Womens Health (Larchmt). 2013;22(10):825-834.
- [Google Scholar]
- What can I do to reduce my risk of ovarian cancer? www.cdc.gov/cancer/ovarian/basic_info/prevention.htm Accessed
- [Google Scholar]
- High-grade serous carcinoma involving fallopian tube, ovary and peritoneum. Surg Pathol Clin. 2011;4(01):375-396.
- [Google Scholar]
- National Cancer Institute. BRCA mutations: cancer risk and genetic testing. www.cancer.gov/about-cancer/causes-prevention/genetics/brca-fact-sheet Accessed
- [Google Scholar]
- American Society of Clinical Oncology policy statement update: genetic and genomic testing for cancer susceptibility. J Clin Oncol. 2015;33(31):3660-3667.
- [Google Scholar]
- Functions of Parylation in DNA damage repair pathways. Genomics Proteomics Bioinformatics. 2016;14(03):131-139.
- [Google Scholar]
- Current development of clinical inhibitors of poly(ADP-ribose) polymerase in oncology. Clin Cancer Res. 2007;13(05):1383-1388.
- [Google Scholar]
- The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol Oncol. 2011;5(04):387-393.
- [Google Scholar]
- PARP inhibitors: synthetic lethality in the clinic. Science. 2017;355:1152-1158. (6330)
- [Google Scholar]
- Trapping of PARP1 and PARP2 by Ccinical PARP inhibitors. Cancer Res. 2012;72(21):5588-5599.
- [Google Scholar]
- PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006;34(21):6170-6182.
- [Google Scholar]
- PARP inhibitors in the management of breast cancer: current data and future prospects. BMC Med. 2015;13:188.
- [Google Scholar]
- Review of mammalian DNA repair and translational implications. J Pharmacol Exp Ther. 2000;295(01):1-9.
- [Google Scholar]
- Perspective on the pipeline of drugs being developed with modulation of DNA damage as a target. Clin Cancer Res. 2010;16(18):4527-4531.
- [Google Scholar]
- Role of BRCA gene dysfunction in breast and ovarian cancer predisposition. Breast Cancer Res. 2000;2(05):324-330.
- [Google Scholar]
- BRCA1 regulates p53-dependent gene expression. Proc Natl Acad Sci U S A. 1998;95(05):2302-2306.
- [Google Scholar]
- BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: a report from the Australian Ovarian Cancer Study Group. J Clin Oncol. 2012;30(21):2654-2663.
- [Google Scholar]
- Somatic mutations in BRCA1 and BRCA2 could expand the number of patients that benefit from poly (ADP ribose) polymerase inhibitors in ovarian cancer. J Clin Oncol. 2010;28(22):3570-3576.
- [Google Scholar]
- Variants of unknown significance in BRCA testing: impact on risk perception, worry, prevention and counseling. Ann Oncol. 2013;24(08):i69-i74.
- [Google Scholar]
- Clinical characteristics of individuals with germline mutations in BRCA1 and BRCA2: analysis of 10,000 individuals. J Clin Oncol. 2002;20(06):1480-1490.
- [Google Scholar]
- Estrogen receptor status in BRCA1- and BRCA2-related breast cancer: the influence of age, grade, and histological type. Clin Cancer Res. 2004;10(06):2029-2034.
- [Google Scholar]
- Tumor suppressor BRCA1 inhibits a breast cancer-associated promoter of the aromatase gene (CYP19) in human adipose stromal cells. Am J Physiol Endocrinol Metab. 2007;292(01):E246.
- [Google Scholar]
- Lynparza. 2:1. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/208558s001lbl.pdf Accessed
- [Google Scholar]
- Spotlight on olaparib in the treatment of BRCA-mutated ovarian cancer: design, development and place in therapy. Drug Des Devel Ther. 2018;12:1501-1509.
- [Google Scholar]
- Functional ex vivo assay to select homologous recombination-deficient breast tumors for PARP inhibitor treatment. Clin Cancer Res. 2014;20(18):4816-4826.
- [Google Scholar]
- Zejula (Niraparib) dosing, indications, interactions, adverse effects, and more. https://reference.medscape.com/drug/zejula-niraparib-1000146 Accessed
- [Google Scholar]
- Jones P, Wilcoxen K, Rowley M, Toniatti C. Niraparib: a poly (ADP-ribose) polymerase (PARP) inhibitor for the treatment of tumors with defective homologous recombination. 2015;58(8):3302–3314
- Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. 2010;376:245-251. (9737)
- [Google Scholar]
- Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361(02):123-134.
- [Google Scholar]
- Final results of a phase 1 study of single-agent veliparib in patient with either BRCA1/2-mutated cancer, platinum-refractory ovarian, or basal-like breast cancer. J Clin Oncol. 2014;32:2570.
- [Google Scholar]
- SOLO2/ENGOT-Ov21 investigators. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017;18(09):1274-1284.
- [Google Scholar]
- Overall survival in patients with platinum-sensitive recurrent serous ovarian cancer receiving olaparib maintenance monotherapy: an updated analysis from a randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Oncol. 2016;17(11):1579-1589.
- [Google Scholar]
- Olaparib as adjuvant treatment in patients with germline BRCA mutated high risk HER2 negative primary breast cancer. OlympiA. 2016;2:1.
- [Google Scholar]
- A phase II evaluation of the potent, highly selective PARP inhibitor veliparib in the treatment of persistent or recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer in patients who carry a germline BRCA1 or BRCA2 mutation - An NRG Oncology/Gynecologic Oncology Group study. Gynecol Oncol. 2015;137(03):386-391.
- [Google Scholar]
- The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trial. Lancet Oncol. 2013;14(09):882-892.
- [Google Scholar]
- Quality of life in patients with recurrent ovarian cancer (OC) treated with niraparib: Results from the ENGOT-OV16/NOVA Trial. Ann Oncol. 2017;28:330-354.
- [Google Scholar]
- A phase III trial of Niraparib versus physician’s choice in HER2 negative, germline BRCA mutation-positive breast cancer patients (BRAVO) https://clinicaltrials.gov/ct2/show/NCT01905592 Updated 2013. Accessed
- [Google Scholar]
- USA National Library of Medicine. A study of Niraparib maintenance treatment in patients with advanced ovarian cancer following response on front-line platinum-based chemotherapy. https://clinicaltrials.gov/ct2/show/NCT02655016 Updated 2016. Accessed Accessed
- [Google Scholar]
- A phase I study of continuous veliparib in combination with IV carboplatin/paclitaxel or IV/IP paclitaxel/cisplatin and bevacizumab in newly diagnosed patients with previously untreated epithelial ovarian, fallopian tube, or primary peritoneal cancer: an NRG oncology/gynaecologic oncology group study. J Clin Oncol. 2015;33:5507.
- [Google Scholar]
- Randomized trial of oral cyclophosphamide and veliparib in high-grade serous ovarian, primary peritoneal, or fallopian tube cancers, or BRCA-mutant ovarian cancer. Clin Cancer Res. 2015;21(07):1574-1582.
- [Google Scholar]
- Phase 1/2 study of oral rucaparib: final phase 1 results. J Clin Oncol. 2014;32:2573.
- [Google Scholar]
- Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): an international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017;18(01):75-87.
- [Google Scholar]
- Phase I trial of olaparib in combination with cisplatin for the treatment of patients with advanced breast, ovarian and other solid tumors. Ann Oncol. 2014;25(08):1656-1663.
- [Google Scholar]
- Phase I trial of the PARP inhibitor veliparib (V) in combination with carboplatin (C) in metastatic breast cancer (MBC) J Clin Oncol. 2014;32:1074.
- [Google Scholar]
- USA National Library of Medicine. A study of MK-4827 in combination with standard chemotherapy in participants with advanced solid tumors (MK-4827–008 AM1). https://clinicaltrials.gov/ct2/show/NCT01110603. Updated 2011. Accessed September 7, 2019
- Phase I trial of the oral PARP inhibitor olaparib in combination with paclitaxel for first- or second-line treatment of patients with metastatic triple-negative breast cancer. Breast Cancer Res. 2013;15(05):R88.
- [Google Scholar]
- Phase I combination study of the PARP inhibitor veliparib plus carboplatin and gemcitabine in patients with advanced ovarian cancer and other solid malignancies. Gynecol Oncol. 2018;148(03):507-514.
- [Google Scholar]
- Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget. 2016;7(12):13587-13598.
- [Google Scholar]
- PARP inhibitor upregulated PD-L1 expression and enhances cancer-associated immunosuppression. Clin Cancer Res. 2017;23(14):3711-3720.
- [Google Scholar]
- Addition of the PARP inhibitor veliparib plus carboplatin or carboplatin alone to standard neoadjuvant chemotherapy in triple-negative breast cancer (BrighTNess): a randomised, phase 3 trial. Lancet Oncol. 2018;19(04):497-509.
- [Google Scholar]
- CTLA-4 blockade synergizes therapeutically with PARP inhibition in BRCA1-deficient ovarian cancer. Cancer Immunol Res. 2015;3(11):1257-1268.
- [Google Scholar]
- The PARP1 inhibitor BMN 673 exhibits immunoregulatory effects in a Brca1(-/-) murine model of ovarian cancer. Biochem Biophys Res Commun. 2015;463(04):551-556.
- [Google Scholar]
- Study of Niraparib in combination with Pembrolizumab (MK-3475) in patients with triple-negative breast cancer or ovarian cancer (TOPACIO) https://clinicaltrials.gov/ct2/show/NCT02657889 Accessed
- [Google Scholar]
- Homologous recombination deficiency (HRD) score predicts response to platinum-containing neoadjuvant chemotherapy in patients with triple-negative breast cancer. Clinical cancer research. 2016;22(15):3764-3773.
- [Google Scholar]
- A Phase 1 trial of the poly(ADP-ribose) polymerase inhibitor olaparib (AZD2281) in combination with the anti-angiogenic cediranib (AZD2171) in recurrent epithelial ovarian or triple-negative breast cancer. Eur J Cancer. 2013;49(14):2972-2978.
- [Google Scholar]
- Combination cediranib and olaparib versus olaparib alone for women with recurrent platinum-sensitive ovarian cancer: a randomised phase 2 study. Lancet Oncol. 2014;15(11):1207-1214.
- [Google Scholar]
- A phase I study of durvalumab (D) in combination with olaparib (O) and cediranib (C) in recurrent women’s cancers. Ann Oncol. 2017;28:122-141.
- [Google Scholar]
- Mechanisms of resistance to therapies targeting BRCA-mutant cancers. Nat Med. 2013;19(11):1381-1388.
- [Google Scholar]
- REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature. 2015;521:541-544. (7553)
- [Google Scholar]
- A phase II trial of the PARP inhibitor veliparib (ABT888) and temozolomide for metastatic breast cancer. J Clin Oncol. 2010;28:1019.
- [Google Scholar]
- Efficacy of the combination of ABT-888 (veliparib) and carboplatin in patients with BRCA-associated breast cancer. J Clin Oncol. 2013;31:1024.
- [Google Scholar]
- USA National Library of Medicine. MK-4827 in combination with pegylated liposomal doxorubicin in participants with advanced solid tumors and ovarian cancer (MK-4827–011) https://clinicaltrials.gov/ct2/show/NCT01227941 Updated 2010. Accessed Accessed
- [Google Scholar]
- Phase I study of the poly(ADP-ribose) polymerase inhibitor, AG014699, in combination with temozolomide in patients with advanced solid tumors. Clin Cancer Res. 2008;14(23):7917-7923.
- [Google Scholar]
- Immune checkpoint inhibitors: A new opportunity in the treatment of ovarian cancer. Int J Mol Sci. 2016;17(07):E1169.
- [Google Scholar]
- HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat Med. 2017;23(04):517-525.
- [Google Scholar]
- Signatures of mutational processes in human cancer. Nature. 2013;500:415-421. (7463)
- [Google Scholar]
- Homologous recombination deficiency (HRD) score predicts response to platinum-containing neoadjuvant chemotherapy in patients with triple-negative breast cancer. Clin Cancer Res. 2016;22(15):3764-3773.
- [Google Scholar]
- Clinicopathological features of homologous recombination-deficient epithelial ovarian cancers: sensitivity to PARP inhibitors, platinum, and survival. Cancer Res. 2012;72(22):5675-5682.
- [Google Scholar]
- Tumor mutation burden forecasts outcome in ovarian cancer with BRCA1 or BRCA2 mutations. PLoS One. 2013;8(11):e80023.
- [Google Scholar]
- Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med. 2016;375(22):2154-2164.
- [Google Scholar]
- BRCA1 and BRCA2 mutations correlate with TP53 abnormalities and presence of immune cell infiltrates in ovarian high-grade serous carcinoma. Mod Pathol. 2012;25(05):740-750.
- [Google Scholar]
- Abstract OT1-03-05: A phase III, randomized, open label, multicenter, controlled trial of niraparib versus physician’s choice in previously-treated, HER2 negative, germline BRCA mutation-positive breast cancer patients. An EORTC-BIG intergroup study (BRAVO study) https://cancerres.aacrjournals.org/content/76/4_Supplement/OT1-03-05 Accessed Accessed
- [Google Scholar]
- Olaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: a randomised phase 2 trial. Lancet Oncol. 2015;16(01):87-97.
- [Google Scholar]
- Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med. 2012;366(15):1382-1392.
- [Google Scholar]
- Genetic and biochemical characterization of multidrug resistance. Pharmacol Ther. 1985;28(01):51-75.
- [Google Scholar]
- Ten-year relative survival for epithelial ovarian cancer. Obstet Gynecol. 2012;120(03):612-618.
- [Google Scholar]
- Addressing the elephant in the room, therapeutic resistance in non-small cell lung cancer, with epigenetic therapies. Oncotarget. 2016;7(26):40781-40791.
- [Google Scholar]
- A partial response to reintroduced chemotherapy in a resistant small cell lung cancer patient after priming with RRx-001. Clin Med Insights Oncol. 2016;10:105-108.
- [Google Scholar]
- Olaparib or cediranib maleate and olaparib compared with standard platinum-based chemotherapy in treating patients with recurrent platinum-sensitive ovarian, fallopian tube, or primary peritoneal cancer. https://clinicaltrials.gov/ct2/show/NCT02446600. NLM identifier: NCT02446600 Accessed
- [Google Scholar]
- PARP inhibitors in ovarian cancer: Sensitivity prediction and resistance mechanisms. J Cell Mol Med. 2019;23(04):2303-2313.
- [Google Scholar]
- Molecular pathways: how can BRCA-mutated tumors become resistant to PARP inhibitors? Clin Cancer Res. 2014;20(03):540-547.
- [Google Scholar]
- Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011;12(09):852-861.
- [Google Scholar]
- In vivo anti-tumor activity of the PARP inhibitor niraparib in homologous recombination deficient and proficient ovarian carcinoma. Gynecol Oncol. 2016;143(02):379-388.
- [Google Scholar]
- Functional restoration of BRCA2 protein by secondary BRCA2 mutations in BRCA2-mutated ovarian carcinoma. Cancer Res. 2009;69(16):6381-6386.
- [Google Scholar]
- Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. J Pathol. 2013;229(03):422-429.
- [Google Scholar]
- Efficacy of chemotherapy in BRCA1/2 mutation carrier ovarian cancer in the setting of PARP inhibitor resistance: a multi-institutional study. Clin Cancer Res. 2013;19(19):5485-5493.
- [Google Scholar]
- Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451:1116-1120. 7182
- [Google Scholar]
- 6-thioguanine selectively kills BRCA2-defective tumors and overcomes PARP inhibitor resistance. Cancer Res. 2010;70(15):6268-6276.
- [Google Scholar]
- Homologous recombination deficiency: exploiting the fundamental vulnerability of ovarian cancer. Cancer Discov. 2015;5(11):1137-1154.
- [Google Scholar]
- A PET imaging agent for evaluating PARP-1 expression in ovarian cancer. J Clin Invest. 2018;128(05):2116-2126.
- [Google Scholar]
- The evolving landscape of predictive biomarkers of response to PARP inhibitors. J Clin Invest. 2018;128(05):1727-1730.
- [Google Scholar]
- A genetic screen using the PiggyBac transposon in haploid cells identifies Parp1 as a mediator of olaparib toxicity. PLoS One. 2013;8(04):e61520.
- [Google Scholar]
- Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015;33(03):244-250.
- [Google Scholar]
- Olaparib monotherapy in patients with advanced relapsed ovarian cancer and a germline BRCA1/2 mutation: a multistudy analysis of response rates and safety. Ann Oncol. 2016;27(06):1013-1019.
- [Google Scholar]
- Phase II, open-label, randomized, multicenter study comparing the efficacy and safety of olaparib, a poly (ADP-ribose) polymerase inhibitor, and pegylated liposomal doxorubicin in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer. J Clin Oncol. 2012;30(04):372-379.
- [Google Scholar]
- Long-term efficacy, tolerability and overall survival in patients with platinum-sensitive, recurrent high-grade serous ovarian cancer treated with maintenance olaparib capsules following response to chemotherapy. Br J Cancer. 2018;119(09):1075-1085.
- [Google Scholar]
- Maintenance olaparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med. 2018;379(26):2495-2505.
- [Google Scholar]
- Incorporation of bevacizumab in the primary treatment of ovarian cancer. N Engl J Med. 2011;365(26):2473-2483.
- [Google Scholar]
- A phase 3 trial of bevacizumab in ovarian cancer. N Engl J Med. 2011;365(26):2484-2496.
- [Google Scholar]
- Standard chemotherapy with or without bevacizumab for women with newly diagnosed ovarian cancer (ICON7): overall survival results of a phase 3 randomised trial. Lancet Oncol. 2015;16(08):928-936.
- [Google Scholar]
- Bevacizumab in ovarian cancer: a critical review of phase III studies. Oncotarget. 2017;8(07):12389-12405.
- [Google Scholar]




